
The U.S. Food and Drug Administration (FDA) is reaffirming its plans to actively regulate certain laboratory developed tests (LDTs) that the FDA has historically treated with enforcement discretion. On June 25, 2024, the FDA released a compliance guide to assist small entities in understanding the requirements applicable to in vitro diagnostic products (IVDs) and LDTs.1 By doing so, the FDA signals to the industry that LDT manufacturers should prepare to comply with FDA regulations and that the FDA is standing behind its controversial final rule (LDT Final Rule), notwithstanding the potential outcome of a looming legal challenge.2
The LDT Final Rule and Phaseout Policy
On May 6, 2024, the FDA issued the LDT Final Rule that makes clear that IVDs are medical devices under the Federal Food, Drug, and Cosmetic Act (FD&C Act), including when the manufacturer of the IVD is a clinical laboratory.3 The FDA considers an LDT to be an IVD intended for clinical use and designed, manufactured, and used within a single clinical laboratory that is certified and meets the regulatory requirements under the Clinical Laboratory Improvement Amendments of 1988 (CLIA) to perform high complexity testing.4 For example, an LDT includes a test intended to be performed on samples taken from the human body (e.g., blood or tissue) for clinical use, including to detect a disease or other condition.5
The FDA also provides in the LDT Final Rule a roadmap for gradually phasing out its general enforcement discretion policy for LDTs. Since 1976, the FDA has exercised enforcement discretion such that it has generally not required most LDTs to comply with medical device regulations related to establishment registration and product listing, reporting of adverse events, current good manufacturing practices or the quality system regulation, and premarket review.6 Under the phaseout policy, the FDA will expect IVDs manufactured by a clinical laboratory to generally fall under the same approach as other IVDs. The FDA claims that such oversight is necessary to account for the significant evolution and proliferation of LDTs, which raises concerns about their safety, effectiveness, and security (e.g., the potential risk of cybersecurity threats that may expose sensitive patient genetic information and results).7
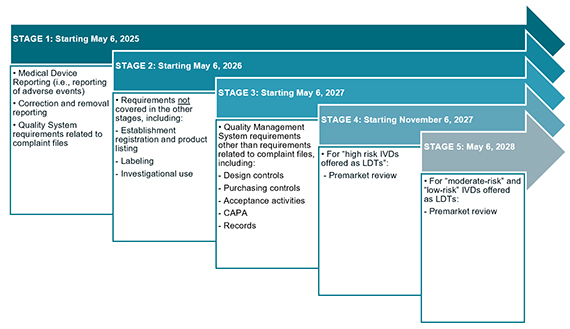
The FDA will stagger the phaseout policy over a four-year period and across five key stages. Notably, the FDA states the phaseout policy will extend beyond the LDTs that have enjoyed the FDA’s exercise of enforcement discretion. The FDA explains the phaseout policy will apply to “IVDs offered as LDTs,” IVDs manufactured and offered as LDTs by clinical laboratories that are certified and meet the regulatory requirements under CLIA to perform high complexity testing and used within such laboratories, even if those IVDs do not satisfy the FDA’s traditional understanding of an LDT because they are not designed, manufactured, and used within a single laboratory.8 The FDA plans the following regulatory requirements for medical devices to take effect for IVDs offered as LDTs starting May 6, 2025, unless exempt.9
Stages of Phaseout Policy
Exemptions from the Phaseout Policy
While the FDA intends to extend greater oversight toward LDTs, the FDA plans to exempt some categories of LDTs from its phaseout policy. The FDA will continue its general enforcement discretion approach (i.e., will generally not enforce any of the medical device regulatory requirements) for the following IVDs because they are “unlikely to pose significant risks” or are “conducted in circumstances that themselves will mitigate the risks”:
- LDTs that share characteristics similar to LDTs that were offered in 1976;
- certain human leukocyte antigen tests for transplantation;
- tests intended solely for forensic (i.e., law enforcement) purposes; and
- LDTs manufactured and performed within the U.S. Department of Defense and Veterans Health Administration for patients treated within such agencies.10
The FDA intends to apply a limited enforcement discretion approach toward other LDTs. The FDA will generally not enforce medical device premarket review requirements for:
- certain LDTs approved by the New York State Department of Health’s Clinical Laboratory Evaluation Program; and
- certain modified versions of another manufacturer’s 510(k)-cleared or De Novo-authorized test.11
Finally, the FDA intends to exercise enforcement discretion and generally not enforce premarket review and Quality System requirements (except for the records requirements under 21 C.F.R. Part 820, Subpart M (records)) for:
- currently marketed IVDs offered as LDTs (i.e., those that were first marketed before May 6, 2024), provided they are not modified after that date or modified only in certain limited ways;
- LDTs manufactured and performed by a laboratory integrated within a healthcare system to meet an unmet need of patients receiving care within the same healthcare system; and
- certain non-molecular antisera LDTs for rare red blood cell antigens for transfusion compatibility.12
Looming Legal Challenge
A month after releasing the LDT Final Rule, the FDA was quickly met with a legal challenge to halt the final rule from taking effect. On May 29, 2024, the American Clinical Laboratory Association (ACLA) and HealthTrackRx, a national trade association representing clinical laboratories and one of its member companies, respectively, filed a complaint in the U.S. District Court for the Eastern District of Texas seeking to set aside and vacate the LDT Final Rule because it violates the Administrative Procedure Act.13 The ACLA makes two principal arguments. First, the ACLA contends that the LDT Final Rule is contrary to law and exceeds the FDA’s statutory authority because it treats LDTs as medical devices subject to regulation under the FD&C Act, when LDTs are instead “professional services” subject to regulation under CLIA.14 Second, the ACLA argues that the LDT Final Rule is arbitrary and capricious and an abuse of discretion because the FDA failed to act with the requirements for reasoned decision-making. The ACLA claims the FDA published the LDT Final Rule without adequately responding to objections to the proposed rule, providing a reasoned justification for the LDT Final Rule, or reasonably explaining its assertion of sweeping regulatory authority.15 The ACLA is asking the court to enter a declaratory judgment against the FDA and an order that vacates and enjoins the FDA from enforcing the LDT Final Rule.16 The ACLA may seek a preliminary injunction to enjoin enforcement of the LDT Final Rule while the case is pending.
On June 28, 2024, in a landmark ruling, the U.S. Supreme Court overruled the Chevron doctrine in Loper Bright Enterprises v. Raimondo,17 which could open the floodgates for more legal challenges to the LDT Final Rule and the FDA’s authority to regulate LDTs as medical devices, as well as other important administrative decisions of the FDA. The pending challenge to the LDT Final Rule may be one of the first FDA cases to be decided under Loper Bright. In light of these uncertainties in the industry, companies that are developing or commercializing LDTs should consider regulatory strategies for mitigating their risks in view of the phaseout policy, such as leveraging clinical data collected through a commercial LDT business model to support a marketing application for FDA premarket review.
Contact Us
For questions regarding the LDT Final Rule, further developments with the phaseout policy, FDA regulatory strategy and risk mitigation considerations for LDTs, IVDs, and other FDA-regulated products, please contact Eva Yin, Jonathan Trinh, or any member of Wilson Sonsini’s FDA regulatory, healthcare, and consumer products practice.
[1] See U.S. Food and Drug Admin., Guidance for Laboratory Manufacturers and FDA Staff, Laboratory Developed Tests: Small Entity Compliance Guide (June 2024), available at https://www.fda.gov/media/179543/download.
[2] See 89 Fed. Reg. 37286, 37286 (May 6, 2024).
[6] Id.; Small Entity Compliance Guide, at 2.
[7] 89 Fed. Reg. at 37289; Small Entity Compliance Guide, at 3. According to the FDA, today’s LDTs: i) are more often used in laboratories outside of the patient’s healthcare setting; ii) are more often run in high volume for large and diverse patient populations; iii) are more commonly manufactured with instruments or components not legally marketed for clinical use yet more frequently used to help guide critical treatment decisions, screen for common diseases, predict personal risk of developing certain diseases, and diagnose serious medical conditions; iv) are manufactured by large laboratories, which run their LDTs on specimens from across the country in large volumes in a single laboratory; and v) increasingly rely on high-tech or complex instrumentation and software to generate results and clinical interpretations. 89 Fed. Reg. at 37289; Small Entity Compliance Guide, at 2; U.S. Food and Drug Admin., Laboratory Developed Tests (updated May 16, 2024), https://www.fda.gov/medical-devices/in-vitro-diagnostics/laboratory-developed-tests.
[8] 89 Fed. Reg. at 37295; Small Entity Compliance Guide, at 4. “IVDs offered as LDTs” do not include IVS manufactured or used outside of a laboratory, including collection devices. 89 Fed. Reg. at 37295 n.21; Small Entity Compliance Guide, at 4.
[9] 89 Fed. Reg. at 37307–11; Small Entity Compliance Guide, at 8–9. In addition to the FDA phaseout timeline, companies should note that, beginning on February 2, 2026, the new Quality Management System Regulation will go into effect, which replaces the existing Quality System regulation under 21 C.F.R. Part 820 and incorporates by reference certain requirements of the International Organization for Standardization (ISO), ISO 13485:2016. See 89 Fed. Reg. 7496 (Feb. 2, 2024).
[10] See 89 Fed. Reg. at 37297–98; Small Entity Compliance Guide, at 5–6.
[11] See 89 Fed. Reg. at 37299–301 and 37310; Small Entity Compliance Guide, at 6.
[12] See 89 Fed. Reg. at 37301–07; Small Entity Compliance Guide, at 7.
[13] Compl. at 1, Am. Clinical Lab. Ass’n v. U.S. Food and Drug Admin., No. 24-cv-00479 (E.D. Tex. May 29, 2024), available at https://www.acla.com/wp-content/uploads/2024/06/ACLA-LDT-Complaint.pdf; Am. Clinical Laboratory Ass’n, Press Release, ACLA CHALLENGES FDA’S FINAL RULE TO REGULATE LABORATORY DEVELOPED TESTING SERVICES AS MEDICAL DEVICES (May 29, 2024), https://www.acla.com/acla-challenges-fdas-final-rule-to-regulate-laboratory-developed-testing-services-as-medical-devices/.
[17] 603 U.S. ___, No. 22-451 (June 28, 2024). The Chevron doctrine, or the Chevron deference, established in 1984 in Chevron U.S.A., Inc. v. Natural Resources Defense Council, Inc., 467 U.S. 837 (1984), has been a cornerstone of administrative law, under which courts have deferred to an agency’s reasonable interpretation of law where the meaning of the law is unclear. Without the Chevron doctrine, many important FDA decisions based on the FDA’s statutory interpretations will be subject to litigation and judicial scrutiny.
