
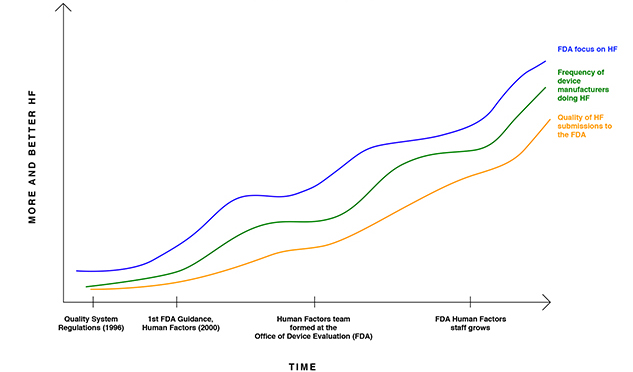
Summer 2017 By Shannon E. Clark, P.E., CEO, UserWise, Inc. According to a recent British Medical Journal research report, the mean rate of death from medical error in U.S. hospitals is estimated to be over 251,000 people per year. Though many medical errors are due to medication errors, patient hand-offs, and issues with hospital processes, other errors are attributable to poor medical device design.1 Human factors engineering plays an essential role in reducing the rate of these avoidable deaths and additional adverse outcomes. The U.S. Food and Drug Administration (FDA) defines human factors engineering as: The application of knowledge about human behavior, abilities, limitations, and other characteristics of medical device users to the design of medical devices including mechanical and software driven user interfaces, systems, tasks, user documentation, and user training to enhance and demonstrate safe and effective use. Overall, the objective of human factors engineering is to minimize or eliminate human error through the design of the medical device. The FDA defines use error to mean: user action or lack of action that was different from that expected by the manufacturer and caused a result that "Human factors" at the FDA is synonymous with usability risk reduction. The focus of usability risk reduction is to design a medical device that minimizes risks related to human error. The errors of focus when reducing usability risks from most devices are usually cognitive in nature (i.e., not ergonomic in nature). The need for usability testing is driven by FDA regulation 21 CFR 820.30(g), which states: Design validation shall ensure that devices conform to defined user needs and intended uses and shall include testing of production units under actual or simulated use conditions. Usability validation testing is one type of design validation, and it includes bringing in end-users to simulate use of the final medical device in a simulated use environment. There is a trend of increasing FDA enforcement of human factors requirements for medical device development and, correspondingly, increased adoption of human factors processes among medical device manufacturers. The FDA shared a graph titled Center Effort on HF/Usability and Industry Response in 2011 (Figure 1). The graph suggests that there was a low level of FDA focus on human factors when the quality system regulations were published in 1996. As a result, very few manufacturers were incorporating human factors processes when the FDA issued its first human factors guidance in 2000. Figure 1: FDA Focus on Human Factors and Industry Response
*Adapted from Human Factors/Usability for Medical Devices: An Historical Perspective, Ron Kaye Office of Device Evaluation, CDRH, Food and Drug Administration, NIST Workshop on Usability and EHR Technology, June 7, 2011. On February 3, 2016, the FDA updated its human factors guidance, Guidance for Industry and FDA Staff – Applying Human Factors and Usability Engineering to Medical Devices (originally published in 2000). The effect of the amended guidance has been rippling quickly through the medical device industry. While manufacturers of higher-risk medical devices began adopting the new human factors processes in 2011 and earlier, many new and established medical device manufacturers continue to scramble to incorporate the new human factors process. The FDA Human Factors Engineering Process The FDA-recommended human factors process can be described in five steps, as depicted in Figure 2. Figure 2: The FDA-Recommended Human Factors Process
Step 1: User Research The goal of user research is to help identify and refine design requirements by understanding the users and the use environments. The focus of user research is on making data-backed assumptions about who the end-users will be and how they will perceive and interact with the product in their context of use. User research helps to identify design requirements by obtaining direct input from intended users. It reveals what people are really thinking and how people really behave through observation at point of use. The researcher evaluates environmental, social, and motivational aspects of the design. User research can include shadowing doctors, nurses, and technicians in various hospital settings and/or conducting one-on-one interviews to reveal user needs. For home-use medical devices, user research is particularly valuable for defining where the device may be used (e.g., Will they keep their device at their bedside or on a wet surface on their bathroom sink? What level of water ingress testing should we consider conducting?). Step 2: Use-Related Risk Analysis A use-related risk analysis is performed to:
The use-related risk analysis is a powerful tool to prioritize design efforts and tackle the most serious use errors first. Various use errors are prioritized according to how serious their consequences are, how frequently they occur, and how easily they can be detected by end-users. It is best to eliminate the given use errors through the design of the device. If it is not possible to eliminate a use error through design, the following options are pursued, in order of preference:2 1. Guard against the use error Step 3: Iterative Prototyping and Usability Testing Usability testing is conducted with early-stage prototypes to reveal the prototypes strengths, weaknesses, and potential use errors. Usability testing includes observing intended users interaction with the device to reveal potential use errors. Subjective feedback on the design is also collected from the usability study participants, but observation of end-users is usually the primary method for collecting data. Multiple early-stage usability studies—called formative usability testing—are usually conducted. In response to each study, rapid modifications are made to the device design in an effort to improve the usability and reduce use errors. It is an iterative process consisting of three stages:
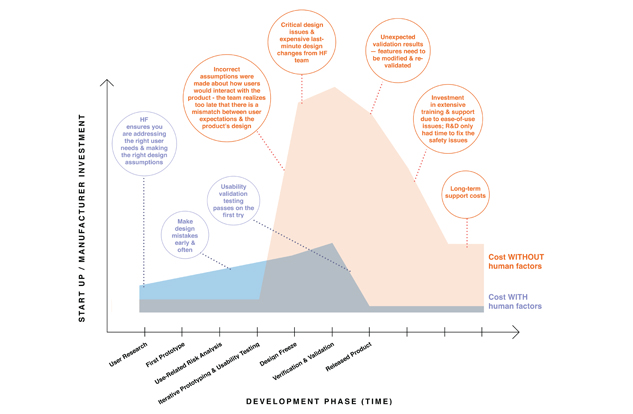
These three stages are repeated until results from formative usability testing show that the design is ideal or that the use errors are minimized. Investing in iterative prototyping and usability testing can save time and money in the long run. The process allows a company to take shortcuts toward an optimized product by allowing imperfect designs to fail more quickly. It is expensive to invest in tooling and manufacturing of final prototypes, and the human factors engineering process facilitates obtaining usability data using low-fidelity prototypes. By conducting multiple usability studies and refining the product cheaply and early on, the development team can progress to the next development phases with greater confidence. Figure 3: Cost of Medical Device Development With and Without Human Factors
Step 4: Usability Validation At the end of the development process, usability validation examines user interactions with the device user interface to identify use errors that could result in serious harm. Usability validation also demonstrates that mitigations in the use-related risk analysis are sufficient to minimize use errors. Per FDA requirements, usability validation testing must include the following:
The results of the usability validation study are used to demonstrate the safety and efficacy of the device with respect to usability. Step 5: Human Factors Submission/Compliance A human factors submission report is prepared at the end of the human factors process. This report describes the full human factors process and explains how use errors were minimized. A human factors engineering submission report is usually required for a pre-market approval (PMA) submission, and the FDA reserves the right to request the human factors engineering report for other types of submissions as well (e.g., 510(k)). UserWise recommends always submitting a human factors engineering submission report in order to expedite and streamline the FDAs review of a submission. For compliance outside of the U.S., it is necessary to assemble a usability engineering file and compliance checklist for IEC 62366-1:2015, Medical devices – Part 1: Application of usability engineering to medical devices. Tips on Human Factors Here are some common human factors pitfalls that UserWise has seen companies encounter:
Conclusion Usability testing is a rigorous process that has become increasingly important for obtaining a 510(k) clearance or a premarket approval. Planning and testing during the early stages of product development can yield great benefits and efficiency in bringing a product to market. The human factors process can save huge amounts of research and development time and money, as well as minimize delays during an FDA submission, reduce the risk of product recalls, and reduce on-market training and maintenance costs.
Before founding UserWise in 2015, Shannon was a human factors engineer at Intuitive Surgical and Abbott Laboratories. She graduated in 2010 from UCLA with a B.S. in mechanical engineering and a technical breadth in technology management. Additionally, Shannon is a Certified Professional Industrial Engineer, holds two patents, and has written and published three books. She can be reached at Shannon.Clark@UserWiseConsulting.com. About UserWise
1 Martin A. Makary and Michael Daniel, "Medical error—the third leading cause of death in the US," 353 British Medical Journal i2139, 2016. 2 Content from ISO 14971:2007. An Interview with Justin Klein of New Enterprise Associates
Tell us about NEA. Whats the firms overall mission and how does NEA try to differentiate itself across its core markets? NEA is a classically constructed venture capital firm. Were going to celebrate our 40th anniversary this year, making us one of the oldest and—because of our firm size and strategies—one of the largest, most active venture capital firms across all sectors. Technology innovation, which broadly includes categories like consumer or enterprise-oriented technologies and electronics, makes up a substantial portion of where NEA invests. And healthcare is the other major category in which we focus our efforts. Within each NEA investment fund, which we tend to raise every 2.5 to 3.5 years in regular cycles, we're committing about a third of our dollars to the healthcare space, which includes biopharma therapeutics, medical devices, and healthtech, as well as healthcare services and healthcare IT. One of the things we prioritize as a capital partner to entrepreneurs is being in a position to actively guide our portfolio companies to expand their market opportunities and scale with our capital and other resources over time. We raise some of the largest funds in the industry, and we believe that being able to invest capital at scale allows us to be an entrepreneurs partner throughout his or her companys lifetime, from seed and Series A stages all the way through growth equity, and potentially as they go public and beyond. What weve found is that in almost all of our sectors, it is increasingly capital-efficient to start a business and demonstrate early traction in multiple markets. But to really scale, a business continues to take resources, and NEA strives to be in a position where we can partner with those entrepreneurs early, help them craft and further expand their vision, and then be their lead financial partner for every step of the journey. What are you looking for in your portfolio companies in terms of unique qualities or traits of success, particularly in the healthcare space? In the healthcare space, were most focused on investing in companies that present open-ended business opportunities, such as standalone businesses that could go public and self-finance over time, or those that become coveted acquisition candidates for some of the larger players in the industry. We like to build companies and franchise opportunities predicated on solutions that address significant unmet clinical needs, and do so at reduced costs. I think the pairing of those phrases is important. Its something we've been focused on for our entire history. To be something big, we believe a company really has to demonstrate evidence to convince all stakeholders to adopt new technologies or new ways of delivering healthcare. Of course, we also look at other things like the nature of the unmet need, the clinical development hurdles, the regulatory path, reimbursement/payment structures, and the go-to-market opportunities to commercialize something. These are all critical elements. Thematically, we try to stay open-minded, focusing on different subsectors, whether its therapeutics, devices, or services, and over time we migrate toward the larger, open-ended opportunities in each category. In the last few years, youve been a part of some of the largest-ever exits for venture-backed medical device companies. Looking forward, what opportunities do you see and what concerns you most about the current healthcare market? Broadly, we remain very enthusiastic about investing in healthcare. We try to be mindful of things like economic and political cycles that could affect our portfolio companies and therefore our investments. If possible, we try to identify long-term secular trends that we think our companies will succeed in, regardless of some of the shorter-term market or political cycles. Our system faces real challenges in terms of the affordability of—and access to—healthcare. We expect that to continue to be a very hot topic. Ultimately though, healthcare is one of the most important dimensions of a persons life. Its close to 20 percent of our GDP, and the opportunity for technology to improve clinical outcomes or reduce costs still remains fairly open-ended. We want to be careful not to invest in entities that bear significant political risks, where opinions about how to do things fall in or out of favor, which could completely derail an investment opportunity. But we do believe there are some durable trends that allow us to invest in a number of these companies and to support them from their earliest stages all the way through to being mature businesses. Do you feel like the healthcare industry is on an upward trend? Do you envision more activity in the next few years, or at least in 2017? Were coming out of a significant bull market in the biopharma space as of a couple of years ago. Of course, there also have been some pullbacks along the way, but most people believe there can be a relatively healthy IPO window this year in multiple healthcare subsectors. Public market investors continue to look for growth opportunities in their portfolios, and strategic acquirers need to find revenue growth opportunities in new businesses to expand their markets, particularly after a period of consolidation among a lot of the big pharma and medtech companies. And were seeing financing environments and acquisition/IPO discussions look fairly positive across all of our categories. So, we think it's going to continue to be a fairly healthy time in the ecosystem. In January 2016, you gave an interview at the J.P. Morgan Healthcare conference where you underscored NEA's commitment to investing in early-stage companies and, specifically in your case, early-stage medical device companies. Can you offer some insights into NEA's reasoning and commitment to early-stage companies? I recently did a quick tabulation of our medical device investment activity in our last two funds, and between those, we made 13 new investments, nine of which were at the Series A or seed stage, including companies that we seeded in incubators. Those numbers would probably surprise most folks, because overall, the medtech venture market has shifted away from pre-regulatory approval or pre-data-stage medical device companies since around 2008. On the contrary, we have deliberately tried to embrace that stage of investment because, one, there are still a lot of opportunities and, frankly, there is less competition from other investors investing in those deals. And two, at a high level, we're trying to invest in the parts of the medtech ecosystem where we as investors and our start-up companies have some competitive advantage. There can be merits to a late-stage investment focus, but its also important to recognize that there are competencies that big, established companies have in these channels, like commercial distribution or manufacturing, that are difficult for a start-up to compete with. Where our companies excel is in identifying unmet needs, developing innovative products that have IP protection, and executing on a development plan that generates evidence for the FDA, payers, patients, and physicians to really embrace things and bring them to market. Some of our peers have moved away from earlier-stage investing, but I don't think thats irrational. From 2008 to 2012, particularly for medical devices, there were a lot of headwinds, particularly around the U.S. regulatory process. Although weve seen the regulatory climate become much more reasonable and predictable in recent years, that era was so taxing for investors and their portfolio companies that its hard to stomach re-testing earlier-stage investment where capital requirements and timelines were extended pretty significantly, almost beyond the reach of a lot of our peers. We are intentional in our strategy to raise relatively large funds, which gives us the ability to sustain our commitment to companies over the long term, and gives them the opportunity to complete the mission. When we invest in early-stage companies, we try to be very thoughtful about the total capital requirement and syndicate formation. I think maybe 10 to 15 years ago, we might have taken on some Series A innovations or technologies that would have required a series of multiple de-risking financings over time, whether its validating technology development, clinical evidence, regulatory approval, reimbursement, or commercial traction. There are probably fewer of those types of opportunities were willing to step up for. We try to find spaces where our companies are in an overall strong position to execute on a plan that answers really hard stakeholder questions relatively early in the process of building that company or funding that program. What are some of the key events that you look forward to attending each year? Are there any new conferences that you're eyeing? Annually, there are a handful of events I try to attend that are fairly spaced out during the course of the year. It probably starts in January with the J.P. Morgan Healthcare Conference, which is kind of the annual must attend. There are a couple of conferences in the spring and early summer, whether it's WSGRs Medical Device Conference, the MedTech Investing Conference in Minneapolis, or Piper Jaffrays annual conference. Then, in the fall, there are some different events that investment banks or other industry groups put together, and those are a great way to keep in regular touch with people. Throughout the year, I typically attend a handful of conferences that focus on clinical areas where we have active portfolio companies, such as cardiovascular disease, interventional pain, or personalized medicine. And sometimes I attend conferences that overlap the due diligence were doing on a new space. Outside of the U.S., are there any particular markets that you or your companies are most interested in? As a firm, NEA is certainly global in its reach. Our interest in start-up companies, as well as the markets where they'll bring their innovations, is global. We have a very active investment practice in Asia, largely on the tech side, though weve made some select healthcare investments there over time. More recently, we've expanded our investment practice to include more opportunities in Europe. One of the companies I'm involved with is called FIRE1 (Foundry Ireland), which is an Ireland-domiciled medical device incubator that we funded in partnership with the Foundry, Lightstone Ventures, and Medtronic. Since creating the incubator, weve advanced the program to include an outstanding senior management team that's based on the ground in Ireland, and we're actively building the company there. Some other examples of investments in our biopharma practice have been companies coming out of Western Europe and the UK, including Adaptimmune in the immuno-oncology space, NightstaRx in the gene-therapy space, and CRISPR Therapeutics in the gene-editing space. Overall, something like 90 percent of our dollars are committed to U.S.-domiciled companies. But we recognize that terrific innovation is happening all over the globe, and were comfortable with backing teams based in those countries. We're working with them to build our companies across the Atlantic, sometimes opening offices and/or taking them public in the United States, and in other cases growing them for the long term, regardless of borders. Do you have any advice for entrepreneurs who are looking to work with NEA or who may be trying to start a company for the first time? First, I think its encouraging that the past five years have been a fantastic time to start a company and raise capital for that company, whether it's in tech or healthcare. There's a lot of fundamental innovation happening in all sectors of the venture ecosystem thats creating tremendous opportunities for new businesses. We like to see entrepreneurs who are passionate about an area where they have a lot of deep experience. And in general, we try to support them, recognizing that their time is the most precious thing that any of us have to commit to one of these ventures. So, if it's an entrepreneur that really knows their space well and theyve identified a problem and developed a technology-driven solution that we share an interest in, we'd love to talk to them as early as possible in the company's formation process. Whether or not we choose to invest can be affected by a variety of different considerations, but we look for opportunities to get involved where we can make an impact on that company's trajectory. That may mean funding them with the right amount of capital, or it may mean helping them set a vision that aims for something bigger or more expansive than they would have otherwise if they hadn't had that conversation. It's often the case that we meet entrepreneurs but may not invest for three or four years. But along the way, we're able to track their progress and help provide introductions to folks who may join their team, or we may introduce other investors who get involved earlier than we do. Then, at the right opportunity, we'll sign up to lead a financing and, once we do, we are fully committed to them. Having those early conversations during that relationship-building process is fundamental, because these can be very durable partnerships. It's rarely an 18-month relationship; usually its three, five, seven, or even 10 years, and hopefully what comes out of it is interest in doing it again. Around 60 percent or more of our investment opportunities are introduced to us through entrepreneurs or folks that we worked with in the past, and if we had a great experience together, we'd love to find that next venture to do it again and again. I think that touches one last point. You've been an investor for quite some time now. What would you say the biggest differences are between being an investor now and being an investor when you first started? From a personal perspective, this is my 11th year of investing and I've had the benefit of being part of NEA and working with some fantastic folks who came before me in our medical technology practice. I started at NEA as an associate, where I was entirely supporting other partners. Today I'm proud to be on a dozen boards and am actively trying to grow our medical device and healthcare technology investment practice with my colleagues in the service of our companies and our industry. With board responsibilities and other leadership opportunities outside of the firm, I've only become busier over time, which is great. It's been a fantastic experience. In each of these investing climates, the markets move in cycles, whether it's related to politics or the economy, and theres always something to learn or figure out how to do better. It could be solving some sort of complicated financing or M&A transaction, or creating investment opportunities that wouldnt otherwise exist. There are always new, creative ways to do this job better and be a better partner or entrepreneur. So, I don't know whether there have been dramatic differences from my first day to yesterday, but it's a continual process thats been a lot of fun. Justin Klein joined NEA in 2006 and is a partner on the healthcare team. Justin focuses on medical device, healthcare technology, and biopharmaceutical company investments. He serves as a director of Advanced Cardiac Therapeutics, Cartiva, ChromaCode, FIRE1, Intact Vascular, Personal Genome Diagnostics, PhaseBio Pharmaceuticals, Relievant Medsystems, Senseonics (NYSE: SENS), VertiFlex, Vesper Medical, and VytronUS. Justins past board memberships and investments include CV Ingenuity (acquired by Covidien), Nevro (NYSE: NVRO), Topera (acquired by Abbott), TriVascular (NASDAQ: TRIV, acquired by Endologix), and Ulthera (acquired by Merz). He is also a member of the advisory boards for Dukes Innovation & Entrepreneurship Initiative, the Johns Hopkins Center for Bioengineering Innovation & Design, and the National Venture Capital Associations Medical Industry Group and its Medical Innovation and Competitiveness Coalition (MedIC), as well as a member of AdvaMed's Business Development Committee. Prior to NEA, Justin worked for the Duke University Health System—reporting directly to the hospital CEO on health system strategy, finance, and clinical service unit operations—as Duke built one of the nation's first and largest healthcare integrated delivery systems. Justin concurrently earned his M.D. from the Duke University School of Medicine and his J.D. from Harvard Law School. He has also served as a member of the board of trustees of Duke University, where he earned his A.B. in economics and his B.S. in biological anthropology and anatomy. Life Sciences Venture Financings for WSGR Clients By Scott Murano, Partner (Palo Alto)
The data demonstrates that venture financing activity increased during the second half of 2016 compared to the first half of 2016 with respect to the total amount raised and the number of closings. Specifically, the total amount raised across all industry segments increased 22.2 percent from the first half of 2016 to the second half, from $847.05 million to $1,034.83 million, while the number of closings across all industry segments increased 5.7 percent, from 106 closings to 112 closings. Notably, the industry segment with the largest number of closings—medical devices and equipment—experienced a slight decrease in number of closings, but an increase in total amount raised during the second half of 2016 compared to the first half. Specifically, the number of closings in medical devices and equipment decreased 2.1 percent, from 48 closings to 47 closings, but the total amount raised increased 4 percent, from $309.39 million to $321.76 million. The industry segment with the second-largest number of closings—biopharmaceuticals—experienced an increase in number of closings, but a decrease in total amount raised during the second half of 2016 compared to the first half. Specifically, the number of biopharmaceuticals closings increased 6.5 percent, from 31 closings to 33 closings, while the total amount raised decreased 11.8 percent, from $420.39 million to $370.91 million. Meanwhile, diagnostics, the industry segment with the third-largest number of closings during the second half of 2016, experienced increases in both number of closings and total amount raised; the number of closings increased 37.5 percent, from 8 closings to 11 closings, while the total amount raised increased 160.2 percent, from $37.84 million to $98.45 million. All remaining industry segments (in descending order of 2H 2016 number of closings)—digital health, healthcare services, and genomics—were flat or up in number of closings and up in total amount raised during the second half of 2016 compared to the first half. In addition, our data suggests that Series A and Series B financing activity compared to bridge financings and Series C and later equity financings increased during the second half of 2016 compared to the first half. The number of Series A closings as a percentage of all closings increased from 31.8 percent to 41.1 percent, while the number of Series B closings as a percentage of all closings increased from 15.9 percent to 17 percent. Offsetting those gains, bridge financing and Series C and later financing activity relative to all other financings decreased during the second half of 2016. The number of bridge financing closings as a percentage of all closings decreased from 31.8 percent to 26.8 percent, while the number of Series C and later financing closings as a percentage of all closings decreased from 15 percent to 10.7 percent. Average pre-money valuations for life sciences companies increased for Series A financings and Series C and later financings, but decreased for Series B financings during the second half of 2016 compared to the first half. The average pre-money valuation for Series A financings increased 70.1 percent, from $10.86 million to $18.47 million; the average pre-money valuation for Series B financings decreased 58.5 percent, from $105.2 million to $43.65 million; and the average pre-money valuation for Series C and later financings increased 18.6 percent, from $120.97 million to $143.45 million. Other data taken from transactions in which all firm clients participated in the second half of 2016 suggests that life sciences is tied with services as the second-most attractive industry for investment. During that period, life sciences (as well as services) accounted for 24 percent of total funds raised by our clients, while the software industry—traditionally the most popular industry for investment—accounted for 29 percent of total funds raised. Overall, the data indicates that access to venture capital for the life sciences industry increased from the first half of 2016 to the second half. It is also worth noting that financing activity during the first half of 2016 had increased significantly over the second half of 2015, and the second half of 2015 had increased over the first half of 2015—so the second half of 2016 represents the third consecutive six-month period of improved financing activity. Moreover, the second half of 2016 represents the second consecutive six-month period of improved financing activity at the Series A stage in terms of number of closings. The second half of 2016 also saw an increase in pre-money valuations for Series A financings, unlike the prior six-month period, which witnessed an increase in number of Series A closings but a decrease in pre-money valuations. This suggests that companies are moving into a greater position of leverage at the Series A stage, as there are more Series A deals getting done and at relatively higher pre-money valuations.
Patenting MedTech with Software – An Update for Inventors By John Shimmick (Associate, Palo Alto) and Charlie Hagadorn (Associate, Seattle) MedTech includes traditional medical devices and smart devices brought about by the tech revolution. From surgical robotics to apps for smart phones, smart devices and connectivity have forever changed the way we think of healthcare and how it is delivered. Examples of computer-based medical devices include surgical robotic systems and lasers used for LASIK surgery. Additional examples include smart patches worn by patients for remote monitoring and 3D scanners used to plan orthodontic treatment. Even an iPhone programmed with the right app can transform a smart phone into a healthcare instrument.
It is important to note that software can transform old or existing hardware into a new invention. For this reason, an update on recent case law and strategies for claiming MedTech software in the United States may be of interest to inventors.
The Good News: Software Is Still Patentable The United States requires that subject matter recited in the claimed invention be patent eligible. In particular, 35 USC § 101 defines patent-eligible subject matter as any new and useful process, machine, manufacture, or composition of matter, or any new and useful improvement thereof. The Supreme Court has created judicial exceptions that preclude patentability under § 101 and held that laws of nature, natural phenomena, and abstract ideas do not qualify as patent-eligible subject matter. In recent years, several cases in the United States have cut back on the extent to which software can be patented because the claims at issue failed to recite patent-eligible subject matter.1 The good news, however, is that software is still patentable in the United States, provided that the claims are not directed to a judicial exception.2 Under Alice, courts will apply a two-part test. The first step is to determine whether the claim at issue is directed to a judicial exception such as an abstract idea. In step two, the court will consider whether the claims contain an inventive concept sufficient to transform the nature of the claim into a patent-eligible application.3 In assessing patentability under § 101, it is important that the court not oversimply the claims.4 In McRO, the claims were directed to automated animation of lip synchronization to sounds. In particular, the claims were limited to rules that evaluate subsequences consisting of multiple sequential phonemes. These claims were held not to be directed to an abstract idea.5 If claims are directed to an abstract idea, the claims can still be found patentable, so long as they do not preempt the field. In general, courts will look at the technical problem being solved and the solution presented with the claimed invention. In Enfish, the claims were directed to a self-referential table that was a specific type of data structure designed to improve the way a computer stores and retrieves data in a memory table feature. These claims were viewed as a particular implementation of a solution to a problem in the software arts, and were held not to be directed to an abstract idea.6 Some keys to success for software MedTech patents include emphasizing that the claimed subject matter:
MedTech IP features that can help secure patent eligibility include:
U.S. Patent Office Subject-Matter Eligibility Resources Over the past few years, the U.S. Patent Office has produced guidelines and other resources for their examiners with respect to subject-matter eligibility under § 101 and related case law. The evolving guidelines address how examiners should formulate their subject-matter eligibility rejections under § 101 and how examiners should evaluate an applicants response to such rejections. In addition, the U.S. Patent Office also compiles regularly updated summaries of § 101 subject-matter eligibility decisions from the U.S. District Courts, the Court of Appeals for the Federal Circuit, and the United States Supreme Court. The U.S. Patent Office subject-matter eligibility page is available at: https://www.uspto.gov/patent/laws-and-regulations/examination-policy/subject-matter-eligibility. If You Have a Killer App, Consider Patenting It In general, the U.S. Patent Office will give patentable weight to software instructions that are stored on a tangible medium, such as an app. Given that software is patentable, an app that transforms a smart phone into more than a mere phone is still patentable, provided that the statutory requirements are met. In general, drafting the patent application and claims to address the technical problem solved by the software app can help with patentability, because the courts and the U.S. Patent Office often look to this when evaluating whether the claims recite patent-eligible subject matter as noted above. The claims can be written to cover the tangible medium that stores the software instructions of the app. These claims effectively cover the app that someone downloads onto their phone. For example, the claims can be directed to the software instructions of the app that control the local device and handle the processing and display of data. Design Patents Can Protect Unique Devices and Graphic User Interfaces (GUIs) The user display and interface that allows the user to interact with data are areas that may be patentable as well. Where a MedTech application includes a novel and non-obvious device or sensor and GUI, or way of presenting, summarizing, or formatting information, consider supplementing the utility patent application with a design patent application. In general terms, a utility patent protects the way an article is used and works, while a design patent protects the way an article looks (the so-called ornamental aspects). The ornamental appearance for an article includes its shape or configuration, along with the surface ornamentation applied to the article. Both design and utility patents may be obtained on an article if the invention resides both in its utility and ornamental appearance. U.S. patent law has evolved to allow the protection of the visual appearance of a GUI as surface ornamentation on the screen of a monitor or smartphone. This was first announced in Ex Parte Strijland, 26 USPQ 2d 1259 (BPAI 1992). New and non-obvious icons associated with GUIs are protectable via design patents. New and non-obvious aspects of the layout of the GUI, including the specific location of each element and even animations, are also protectable. Design patents have other additional benefits:
Additional Strategies for Protecting MedTech Many approaches can work, depending on the nature of the technology and the invention. In general, it is helpful to describe and claim the invention from several perspectives, including the local device and the remote server. For each of these, it can be helpful to have device claims, method claims, and software claims (i.e., tangible medium claims). For example, the hardware that is used locally is often patentable. Examples of patentable hardware include specific tools used for surgical robotics, stents, and balloons that are used to treat patients. The general requirements for such inventions include novelty, non-obviousness, and utility. The system that is used locally, including the software, can also be patented, for example with a combination of hardware and software. For software inventions, claims can often be written to cover how data is processed in the cloud (e.g., the server). In many instances, it can be helpful to have a hook between the server and the hardware. For example, if there is any special data from the local device that is being sent to the server, claims directed to the receipt and processing of this data with the server can provide useful points of distinction over the prior art. To learn more, please contact Mike Hostetler, Sabrina Poulos, Mike Rosato, Doug Portnow, Jim Heslin, John Shimmick, Scott Burkette, Charlie Hagadorn, or Peter Eng.
1 See, e.g., Bilski v. Kappos, 130 S. Ct. 3218, Supreme Court (2010); Mayo Collaborative v. Prometheus Labs., 132 S. Ct. 1289, Supreme Court (2012); Alice Corp. Pty. Ltd. v. CLS Bank Intern., 134 S. Ct. 2347, Supreme Court (2014). 2 See, e.g., DdR Holdings, LLC v. Hotels. Com, LP, 773 F. 3d 1245, Federal Circuit (2014); ENFISH, LLC v. Microsoft Corp., 822 F. 3d 1327, Federal Circuit (2016); McRO, Inc. v. Bandai Namco Games America Inc., 837 F. 3d 1299, Federal Circuit (2016). 5 McRO, 837 F. 3d at 13133 at 1316. 6 Enfish, 822 F. 3d 1327 at 1339. The Serious and Immense Impact of a Medical Device Hack By David Hoffmeister (Partner, Palo Alto), Vern Norviel (Partner, San Francisco, San Diego, and Boston), Mark Solakian (Partner, Boston), Lou Lieto (Partner, Boston), Lydia Parnes (Partner, Washington, D.C.), Lawrence Perrone (Of Counsel, Washington, D.C.), Wendell Bartnick (Associate, Austin), Jennifer Fang (Associate, Boston), Prashant Girinath (Associate, Boston), Jake Gatof (Associate, Boston), and Charles Andres (Associate, Washington, D.C.) The Muddy Waters report was largely based on analysis conducted by the cybersecurity company MedSec Holdings Inc. MedSec Chief Executive Officer Justine Bone suggested that St. Judes products had an astounding level of problems, including lack of encryption and authentication between devices, which could allow hackers to tap into implanted devices.3 MedSec had negotiated compensation tied to the success of Muddy Waters trade position, and Ms. Bone stated that partnering with Muddy Waters was the most powerful way to inflict pain on St. Jude for what she called its negligent level of attention to cybersecurity.4 At the time of the Muddy Waters report, St. Jude was in the process of being acquired by Abbott Laboratories for $25 billion. St. Jude shareholders were slated to receive, for each share of St. Jude common stock held, $46.75 in cash and 0.8708 shares of Abbott common stock, representing about $85 per St. Jude share, by the end of the year. In contrast, upon release of the Muddy Waters report, St. Jude stock closed at $77.82, well below the deal value, leading analysts to speculate about the prospect of the acquisition by Abbott. In response, St. Jude filed suit in the U.S. District Court for the District of Minnesota against Muddy Waters and MedSec, claiming that the allegations of cybersecurity vulnerabilities are false. St. Jude further alleged that the two companies used false and misleading tactics to scare patients, drop share prices, and make cash on the side as a result. St. Jude also released a rebuttal report stating that the researchers at MedSec used flawed test methodology on outdated software, demonstrating a lack of understanding of medical device technology.5 As the case has proceeded, Muddy Waters has released additional videos and expert reports elaborating on its allegations. Abbotts deal with St. Jude recently closed, and the company has continued to assert that these allegations are exaggerated and untrue.6 In this article, we explore select ramifications of a medical device hack, and provide some suggested practices for companies that offer medical devices to the public. The Regulatory Landscape Companies that manufacture and sell medical devices to the public face a complex regulatory landscape. A host of different government agencies enforce laws that impose obligations on medical device manufacturers whose devices gather, store, or transmit information. HIPAA For example, the Health Insurance Portability and Accountability regulations (HIPAA rules) issued and enforced by the Department of Health and Human Services (HHS) govern the privacy and security of protected health information (PHI).7 The HIPAA rules require implementation of reasonable and appropriate administrative, physical, technical, and organizational data security safeguards, including data security risk assessments, and ongoing risk management efforts to reduce cyber risks and vulnerabilities. Compliance with the HIPAA rules is mandatory for device manufacturers that collect or transfer PHI.8 Device manufacturers and others that fail to comply with the HIPAA rules may face significant penalties. For example, in August 2016, HHS imposed a $5.55 million penalty in a settlement with Advocate Health Care Network due, in part, to an alleged failure to conduct a data security risk assessment and to implement reasonable physical security measures. In roughly the same timeframe, HHS settled a case against Oregon Health & Science University (OHSU) that included a $2.7 million civil penalty. The case was based on allegations that OHSUs risk assessment did not cover all electronic PHI that it maintained, and that OHSU did not reasonably and appropriately address documented vulnerabilities and risks in a timely manner. These settlements underscore the importance of conducting regular risk assessments, ensuring that the device manufacturers data security mechanisms meet ever-evolving threats, and confirming up-to-date HIPAA compliance. The FTC In addition to the specific rules that govern PHI, the Federal Trade Commission (FTC) has taken a similar approach to data security more generally. Relying on the very broad language in Section 5 of the FTC Act, which prohibits unfair and deceptive acts and practices in or affecting commerce, the FTC has brought over 60 enforcement actions against companies that allegedly failed to maintain adequate data security. Some of these actions were based on allegations that a company engaged in a deceptive practice if it did not have measures in place that matched the public representations it made about its data security efforts.9 Even without an affirmative representation, however, the FTC could challenge a device manufacturers data security practices as unfair if the manufacturer failed to employ reasonable and appropriate measures to prevent unauthorized access to the information it collected. The FTCs enforcement actions, virtually all of which are settlements, require companies to implement and maintain data security programs that contain administrative, technical, and physical safeguards appropriate for the size and complexity of the business and the sensitivity of the personal information collected from or about consumers. Similar to HHS, the FTC expects companies to engage in regular risk assessments. Device manufactures should consider implementing data security plans that meet these standards and should review their public statements, including their privacy policies, to ensure that their practices are consistent with any public commitments. The SEC Public medical device companies should also consider whether a security vulnerability or data breach should be disclosed to investors and, by extension, to the U.S. Securities and Exchange Commission (SEC). The SEC has the authority to investigate possible violations of the federal securities laws, which include failures of public companies to make adequate disclosures, withhold material information, and/or misrepresent to, or mislead, investors.10 In 2011, the SEC issued written guidance to public companies to assist them in assessing what, if any, disclosures should be provided [to shareholders/investors] about cybersecurity matters. The guidance notes that [a]lthough no existing disclosure requirement explicitly refers to cybersecurity risks and cyber incidents, if a public company experiences a material cyberattack it would not be sufficient for the company to merely disclose that a risk of cyber-attacks exists (i.e., via standard risk factors); rather, the public company may be required to disclose specifics regarding the cyber event and its potential costs and consequences. Outside of standard risk factor disclosure, the SEC recommends that companies review other disclosures such as the Managements Discussion and Analysis of Financial Condition and Results of Operations (MD&A), Business, Legal Proceedings, and Financial Statement sections. In 2014, former SEC Commissioner Luis Aguilar publicly stated that cybersecurity is of particular concern to the SEC and that he hoped the disclosures discussed in the 2011 guidance helped investors and public companies to focus and assess cybersecurity issues. Current SEC Chair Mary Jo White has reaffirmed the SECs focus on cybersecurity.11 Of course, the dispositive question in determining whether disclosure is required is whether the cyber-attack/security vulnerability is material to investors. In the recent past, many companies that have suffered large cybersecurity breaches have not reported these in their period or current reports on Form 10-K, 10-Q, or 8-K, and there have been a limited number of SEC enforcement actions for failure to disclose breaches. Increasing scrutiny and public awareness of cyber incidents, however, could lead to a tightening of disclosure standards. Public companies should be careful to ensure proper disclosure. The FDA Finally, medical device companies should also consider the U.S. Food and Drug Administrations (FDAs) role in any medical device hack, especially where the hack could result in harm or death to patients. The FDA regulates medical devices under, for instance, the Medical Device Amendments of 1976, and is keenly concerned with the safety and effectiveness of any medical device. Recognizing that the cybersecurity of connected medical devices could present a growing problem, the FDA issued guidance on post-management security in 2016.12 While the FDAs guidance touches on a number of areas, when evaluating post-market risk, the FDA encourages companies to:
The FDA has enforcement authority over medical device manufacturers. If a medical device: a) has uncontrolled risk, including a cybersecurity risk, to essential clinical performance that b) may reasonably cause serious adverse health consequences or death, then the manufacturer may be in violation of the Federal Food, Drug, and Cosmetic Act (FDCA). FDCA violations may subject the device manufacturer to FDA enforcement actions, which can include the seizure and recall of medical devices. Thus, if a medical device hack endangers the health or safety of patients, the medical device manufacturers should work with the FDA13 to mitigate the hacking-associated risks in an expeditious manner. Companies should be prepared to recall medical devices that contain the vulnerability, re-engineer the medical device or its software to remove the hacking vulnerability, and facilitate communication shut-off of in-use medical devices until, for instance, a vulnerability-mitigating patch can be implemented. Reporting obligations to various agencies of the federal and state governments, and mechanisms for addressing any FDA-mandated action, should be contained in the incident response plan that is prepared and in place ahead of any hack. Plan of Action Medical device hacks can have serious and wide-ranging repercussions: they can endanger patient lives, result in data breaches, materially affect stock prices, sour investor relationships, scuttle ongoing transactions, and tarnish a device manufacturers reputation. Hackers may also attempt to use their ability to hack a device to extract a ransom in exchange for not harming patients relying upon the device, for providing information about how the hack is performed, or for containing or preventing a data breach. To prepare for a possible intrusion, companies whose devices may be subject to hacking should develop an incident response plan. Companies should also create a culture that encourages and enables timely reporting, evaluation, and escalation of reports of a possible hack, regardless of the source. This can be achieved, for example, through comprehensive training of personnel and putting into place appropriate internal reporting mechanisms and structures. In addition, companies should consider reviewing existing internal compliance policies, including those related to whistleblowing, to ensure these are designed to appropriately identify and address reports of information technology and cybersecurity issues. For example, whistleblowers and white hat hackers should have appropriate avenues to report potential cyber vulnerabilities. Incident Response Plan and Team The discovery of a hack is, at minimum, unsettling for any company. Senior managers are faced with making decisions under extreme time pressures, which can significantly impact the business. In making these decisions, senior managers must be able to adjust in response to unfolding events and new information. Manufacturers may also have obligations to notify various government agencies such as the FDA and HHS, as well as affected individuals and their caregivers.14 Managing this effort can be complicated and uncertain, and being prepared is a significant factor in mitigating costs and damages associated with a hack. A key factor in security incident preparedness is developing an incident response plan. Supporting the centrality and importance of an incident response plan, research conducted by the Ponemon Institute shows that failure to have an incident response plan and team in place is a leading factor that can increase the incident costs and damages.15 Therefore, companies should draft, implement, and regularly test their incident response plans.16 Incident response plans typically include detailed instructions for the following:
Having and following an incident response plan helps an organization methodically take the proper steps while responding to an incident. Organizations with a plan will be able to more quickly assess the incident so that they can respond in a timely, cost-efficient, and effective manner. Intellectual Property Considerations Timely fixing or patching over the hack is of paramount importance. But the ability to make hardware or software modifications that mitigate a hacking vulnerability may not simply be a technical problem. Any fix to a devices hardware or software should also not violate intellectual property to which the medical device manufacturer does not have rights. Thus, medical device manufacturers should maximize patent claim scope, strategically leverage licenses, and be aware of the relevant patent landscapes so as to create a buffer that allows for modifications that could be reasonably foreseeable in response to a hack. Other Considerations A medical device hack (or the possibility of a hack) raises diverse considerations beyond those discussed above. While it is not possible to address all of these, we point out three relevant examples as catalysts for further thought. First, if a medical device manufacturer is involved in a transaction to sell the company, it should be careful in ensuring proper disclosure regarding the features and limitations of the medical device and proactively addressing any cybersecurity vulnerabilities to limit post-closing issues. The medical device manufacturer should also carefully consider how risk—in the form of indemnification—should be allocated after the deal closes. Second, disclosure of a hack may put downward pressure on a medical device companys stock. To protect against hostile takeover at a vulnerable point, companies may want to consider implementing appropriate protective actions. Finally, one way to minimize fallout from a hack is to control the narrative, which includes providing thoughtful responses, such as planned changes to address vulnerabilities. Strategic, clear, timely, and honest public relations can help a company weather a hack. Any proposed communication, however, should be evaluated in light of the potential for the communication to be used in a future investor or patient lawsuit. Conclusion With the growth of medical devices that communicate wirelessly, share data, and can be adjusted or turned off remotely, the threat, reach, and potential fallout of hacking will continue to increase. Medical device manufacturers should proactively take steps to minimize the possibility of hacking, and have structures in place—including an incident response plan—to deal with a hack, should it occur.
1 See http://www.muddywatersresearch.com/research/stj/mw-is-short-stj/. 3 See http://www.bloomberg.com/news/articles/2016-08-25/carson-block-takes-on-st-jude-medical-with-claim-of-hack-risk. 5 See http://www.reuters.com/article/us-st-jude-medical-cyber-idUSKCN11129K. 6 Despite the companys assertions that the allegations are exaggerated and untrue, the FDA contacted the company on April 12, 2017, and gave them 15 days to explain how it has addressed cybersecurity concerns. 7 The HHS regulations implementing the privacy and data security provisions of HIPAA are at 45 C.F.R. §§ 160, 164. 8 The protocol that HHS uses in HIPAA compliance audits is available at http://www.hhs.gov/hipaa/for-professionals/compliance-enforcement/audit/protocol/. 9 For example, in January 2016, the FTC investigated and settled a case against Henry Schein Practice Solutions, Inc., for its alleged failure to provide industry-standard encryption of patient information despite advertising that it did so. In re Henry Schein Practice Solutions, Inc., No. C-4575 (May 20, 2016). 10 While the SEC engaging in the regulation of cyber or security events may seem odd, it is not. The underlying facts of such securities violations related to a cyberattack or security vulnerability in a medical device likely do not undermine such authority. See Securities Act of 1933 (Securities Act), Sections 19 & 20, 15 U.S.C. §§ 77s, 77t; Securities Act of 1934 (Exchange Act), Section 21, 15 U.S.C. § 78u. 11 Earlier this year, the SEC hired Chris Hetner as its first Senior Advisor to the Chair for Cybersecurity Policy. 12 See Postmarket Management of Cybersecurity in Medical Devices: Draft Guidance for Industry and Food and Drug Administration Staff, FDA (Jan. 22, 2016), available at: http://www.fda.gov/downloads/MedicalDevices/DeviceRegulationandGuidance/GuidanceDocuments/UCM482022.pdf. 13 Manufacturers regulated by the FDA may be required to report certain vulnerabilities under 21 C.F.R. parts 803, 806, and 1004. 14 FTC v. Wyndham Worldwide Corp, 12-CV-1365 (D. Ariz. 2012) (Complaint). 15 Cost of a Data Breach Study: United States, Ponemon Institute (June 2016). 16 HIPAA requires regulated companies to have an incident response plan. HHS recently reached a settlement with the University of Mississippi Medical Center imposing a monetary penalty of $2.75 million for HIPAA violations, including a failure to implement policies and procedures to address security incidents and a failure to properly notify individuals affected by a data breach. 17 Although this article does not deal with device design and manufacturing issues per se, companies should also consider taking steps to minimize the possibility of a device being hacked by: limiting the communication range of the device, using handshake protocols, making use of sophisticated encryption software, and allowing for external communication with the device to be shut off.
Select Recent Life Sciences Client Highlights
Ninth Circuit Affirms Dismissal of Securities Class Action Against Align Technology
25th Annual Medical Device Conference Phoenix 2017: The Medical Device and Diagnostic Conference for CEOs Biotech Board of Directors and Senior Executives Reception
Click here for a printable version of The Life Sciences Report This communication is provided as a service to our clients and friends and is for informational purposes only. It is not intended to create an attorney-client relationship or constitute an advertisement, a solicitation, or professional advice as to any particular situation. © 2017 Wilson Sonsini Goodrich & Rosati, Professional Corporation |

 Shannon E. Clark is the founder and CEO of UserWise, a consultancy that helps medical device manufacturers and start-ups to design safe and easy-to-use medical devices. The consultants at UserWise conduct usability testing for a variety of medical devices, ranging from surgical robots to home-use injection platforms. UserWise consultants also perform safety assessments to comply with U.S. and international regulations related to human factors.
Shannon E. Clark is the founder and CEO of UserWise, a consultancy that helps medical device manufacturers and start-ups to design safe and easy-to-use medical devices. The consultants at UserWise conduct usability testing for a variety of medical devices, ranging from surgical robots to home-use injection platforms. UserWise consultants also perform safety assessments to comply with U.S. and international regulations related to human factors.  Our mission is to inspire human factors engineering best practices within both Fortune 500 medical device companies and start-ups, and to facilitate the development of usable medical devices. We work with companies to fulfill any and all of the steps in the usability engineering process to facilitate the design of safe and usable medical devices. We offer risk analysis, usability testing, and compliance documentation, as well as corporate trainings and assistance navigating regulatory clearance. To learn more, visit
Our mission is to inspire human factors engineering best practices within both Fortune 500 medical device companies and start-ups, and to facilitate the development of usable medical devices. We work with companies to fulfill any and all of the steps in the usability engineering process to facilitate the design of safe and usable medical devices. We offer risk analysis, usability testing, and compliance documentation, as well as corporate trainings and assistance navigating regulatory clearance. To learn more, visit  Wilson Sonsini Goodrich & Rosati partner James Huie recently sat down with Justin Klein, a partner at New Enterprise Associates (NEA), one of the worlds largest and most active venture capital firms. Among other topics, Justin discussed NEAs mission and commitment to investing in early-stage companies, the current state of the healthcare investment industry, and the advice hed offer to entrepreneurs. Below is a selection of highlights from their discussion.
Wilson Sonsini Goodrich & Rosati partner James Huie recently sat down with Justin Klein, a partner at New Enterprise Associates (NEA), one of the worlds largest and most active venture capital firms. Among other topics, Justin discussed NEAs mission and commitment to investing in early-stage companies, the current state of the healthcare investment industry, and the advice hed offer to entrepreneurs. Below is a selection of highlights from their discussion.
 The smart phone illustrated in the figure to the right is an example of many new MedTech devices. Often there is a local device, such as a smart phone, that has sensors or actuators that interact with the local environment. The local device also could be a spectrometer, a surgical robot, a laser eye surgery system, or a diagnostic instrument, for example. A local processor, such as a processor of the smart phone, is coupled to the local device or sensor within the device that may gather data from the local device and may control the local device. The local device transmits the data to a remote server in the cloud. The remote server can be configured to do much more than merely store data—it can be configured to perform analytics and machine learning, and offer guidance to the local device.
The smart phone illustrated in the figure to the right is an example of many new MedTech devices. Often there is a local device, such as a smart phone, that has sensors or actuators that interact with the local environment. The local device also could be a spectrometer, a surgical robot, a laser eye surgery system, or a diagnostic instrument, for example. A local processor, such as a processor of the smart phone, is coupled to the local device or sensor within the device that may gather data from the local device and may control the local device. The local device transmits the data to a remote server in the cloud. The remote server can be configured to do much more than merely store data—it can be configured to perform analytics and machine learning, and offer guidance to the local device. 
















