
Fall/Winter 2018 Interview with Dr. Daniel Burnett of TheraNova LLC Wilson Sonsini Goodrich & Rosati partner James Huie recently interviewed Dr. Daniel Burnett, president and CEO of TheraNova. TheraNova is an experienced medical device developer with a track record of creating innovative and practical solutions to large markets with unmet needs. During the interview, Dan touches on a number of topics, including TheraNovas mission, what hes learned since starting the company, and factors that are fueling innovation in the medtech sector. He also offers helpful advice to startups and entrepreneurs. James: Tell us about TheraNova and the incubators business plan and overall value proposition. Dan: TheraNova started as an IP holding company when I joined MedVenture Associates, the venture capital firm. MedVenture invested in my first company, but at the time, I also had patents unrelated to that company, so I formed TheraNova to hold them. In 2005, I left MedVenture Associates when I realized I liked building companies more than investing in them. I started to create some structure around TheraNova, moving it beyond being an IP holding company, because it was no longer possible to get a company funded based on just a patent. You needed either some sort of benchtop or preclinical data. The first company I had funded was based on just the patent application and business plan. The second one was based on preclinical data. Every one after that required human clinical data. So we formed TheraNova to provide the level of support needed to get that first in man data. In order to hit this critical milestone we started to bring in engineering expertise, and then clinical expertise. As we spun out more companies, we started to get this critical mass where the portfolio companies could collectively support a patent person, a grant writer, and a regulatory person. We ended up bolting on more and more expertise, and now we essentially have all the expertise in-house that a startup would typically need to outsource with variable results. Other than reimbursement, we have dedicated and highquality in-house personnel in all the key areas. James: Those are valuable resources for your incubating companies and potential spin-offs. At last check, I believe TheraNova had spun off 14 companies that have raised more than $280 million in venture funding. What factors have been important to their success, and what considerations go into deciding whether or not to spin off a company?
We did have one technology fail prior to clinical data. It hadnt taken any outside investor capital, which was good, but we just decided to call it a loss and not pursue it further. It was a breast implant that had an inductive sensor that would signal if the implant was leaking. There were two problems. First, we had a partner lined up, but they were acquired at a point when we were far along in talks. This consolidated potential acquirers and resulted in an undesirable duopoly. Second, there was the introduction of a so-called gummy bear implant, in which the silicone was a cohesive gel that didnt leak. So the market had evolved underneath us and it essentially erased the value proposition for the technology. Beyond that, we simply continue marching through the benchtop, preclinical, and clinical stages, then try to get key opinion leader (KOL) support. Once we have KOL support, plus proof of principle and initial demand, we can start testing the waters with investors. One of the bigger risks for medical device companies these days is financial. So, we test the waters with investors and potential acquirers. If we get interest from both, we fast track the project. James: TheraNova certainly can provide resources that are vertically quite deep. Tell us about the recent partnership with UCSF Surgical Innovations, which we understand is a vehicle for UCSF medtech innovators. Dan: Ive worked with them for a while, but we have not worked together as closely as we are now. That may be because of the partnership announcement, but also because we are increasingly reaching out to them to do more clinical studies and get clinical input for what were doing at TheraNova. Up until recently, TheraNova has had clinical people, but Im the only M.D. on staff, so broadening our clinical expertise and depth of knowledge was important to us. TheraNovas UCSF Surgical Innovations partnership made a lot of sense to me and their faculty directors, Dr. Shuvo Roy and Dr. Hanmin Lee. Ive worked with Dr. Roy extensively as an industry director for the Master of Translational Medicine (MTM) program. Hes the academic director. Ive also started to work more with Dr. Lee, who is a strong force for innovation at UCSF. James: Its exciting to see the collaboration, especially as Mission Bay and UCSF continue to grow, and great to have a partner that knows how to take technology beyond academia. There has been a lot of progress at TheraNova since you started. What are some of the things that youve learned along the way, if you compare your knowledge and understanding then to what you know now? Dan: I learned a lot on the investor front. When I first started, I was really focused on pre-money valuation and trying to make the financing less dilutive. Then, I began to realize that thats not nearly as important as picking the right investor partner—one that has the same goals and similar views on what a successful arc for the company looks like. I also learned that it used to be all about improving outcomes and decreasing morbidity and mortality. Thats all that mattered. If you came up with something that did both things, and you incentivized the physicians or healthcare systems by being profitable for them, that was a victory. Now, increasingly weve been focusing on the triple aim: we look for technologies that improve outcomes, reduce costs, and expand access to care. That was based in part on the MTM program that was started by Andy Grove at Intel. He and Dr. Roy came up with that triple aim for their program, for technologies that they were looking for. We adopted it based on the recognition that if you can develop technologies that do all three things, then you have everybody pushing for you—the Food and Drug Administration, Centers for Medicare & Medicaid Services, hospitals, payors, patients, physicians. Everybody. James: Thats a great perspective because I come across a number of companies that might have one of those three, or maybe two. But then they get tripped up when they start to talk to investors, or even later as acquirers. Whats your view of the medtech sector? Do you see future growth in the near term? Dan: Its interesting. There are a lot of macroeconomic developments taking place right now. The medtech industry is finally turning around. Youre starting to see funds surface, like Medtech Venture Partners, as well as others that are either focused only on medtech or have a medtech component. Obviously, predictive health and the digitalization of medical devices are huge. So, its not just digital health, but also having smarter devices. That includes previously dumb devices—like tubes in the body—that now have some kind of functionality to them. Thats been one of our big thrusts where we have incubated technologies that extract actionable data from commodity devices like urine drainage systems (Potrero Medical), feeding tubes (Gravitas Medical) and PICC lines (Piccolo Medical). We are confident that by generating clinically useful algorithms from proprietary data streams we will have the potential to create the next medtech unicorns. I also think that medicine will become much easier for doctors. For example, many of these technologies will be used as an adjunct in caring for the patient, to alert them of situations that they wouldnt be aware of clinically, but that can be detected by sensors in combination with artificial intelligence. These technologies are processing data streams that would be impossible for a human to take into account. James: Thats quite interesting, and it highlights something thats always been part of medtech: adoption by doctors, physicians, and surgeons. Do you see this new wave of medical devices having similar adoption hurdles? Dan: Thats a good question. I think its going to be similar based on the technology. If you have a technology that is what we call a missionary sell—meaning youre taking a technology that the hospital or doctor has to believe in to do something new or different—then you end up with a market that needs to be convinced. This is especially true if the need your technology is satisfying isnt clearly identified or its not a glaring need for the physician in a hospital system. But if you have a technology that provides actionable data, that is useful and valuable to the physician healthcare system as it is, then you can drive adoption. For example, at Potrero Medical, we originally set out to fix urine output. Along the way, we added a temperature sensor and a pressure sensor to the device, so they now provide urine output with accuracy and frequency that was previously impossible. Although hospitals are buying the device based mainly on the urine output aspect, were also collecting the other two data streams and we think it will help revolutionize ICUs. That is, by tracking those data and using machine learning and analytics, we can detect illnesses in the ICU much earlier. We have already shown this in a study of acute kidney injury that we presented at the Military Health System Research Symposium and expect the correlations to continue to strengthen and broaden. James: Its interesting to see the convergence of technology. I always wonder whether the digital health craze is just the new flavor of the week for Silicon Valley, or if its really representing a pivotal time where we see convergence across industries like medtech, software, big data, AI, and sensors. I also think about whether the new technologies werent available before only because industries werent talking to each other. Dan: I dont think its like the dot-com era where its blowing up bigger than it should, or that were in a bubble. I believe there will be technologies that do revolutionize healthcare within 10 years. That will come about because of what you just said: less expensive sensors, medtech innovations, big data, machine learning, and analytics. And because of the last piece thats going to be required—evidence-based medicine. So if these technologies improve healthcare to the extent that convinces doctors, theyll be adopted more rapidly. Youll see a lot of victories related to devices that help doctors do their job better. James: Its certainly great just having successful technologies brought to market. Another common challenge for start-ups is the tendency to burn through cash and run out of money. In an article you wrote in October 2014, you noted that the industry average for bringing a concept to market was about $31 million. What strategies would you recommend to companies that want to stay lean and implement cost-efficiencies? Dan: The best recommendation I can make is to look for synergies. For us, the biggest source of efficiency is when we have a technology that leverages the work from another technology. So, for example, we have a feeding tube that weve developed where we are already in discussions with potential acquirers prior to an equity financing. We got to our first in man with that device for about $25,000, which is probably close to two orders of magnitude less than the industry average. That was possible because we used the hardware and software library from Potrero Medical and adapted it to provide the readings that we needed for our feeding tube to show that its in the stomach and not in the lung. And weve got a great electrical engineer and software expert who was able to adapt the technology so that we could get these readings, and then, as a physician, I was able to put the tube in myself and show that it was in the right place. So, that was our clinical data. At TheraNova, we have an expression: Instead of first in man, its first in Dan. As long as its not invasive, Ill usually try it out first. James: Thats pretty amazing. Weve really seen you evolve throughout your career, and now youre taking on a mentor role—we know that you joined UCSF as an entrepreneur-in-residence with QB3 and youre an adjunct professor in UCSFs bioengineering department. Do you have any advice for young companies and potential entrepreneurs when theyre starting their careers with their companies? Dan: Dont focus on the valuation and be careful when picking the firm thats investing in you. Its more important to pick the investor that feels right for the company and has a similar mindset as you. So, my advice is that, when you pick your investor, think of it as getting married because the two of you—the founders and the investors—will be the parents of that spin-out, and if you are in sync in terms of what you want for that company, things will be better in good times and in bad. James: Just seeing the community that youre building there, does TheraNova have a mission statement? I see everyone working together, and I feel like they have a common ethos. Dan: Yes, I think having an ethos of focusing on impact and not just financial return is critical to what we do and critical to us being able to hire talent. We focus on reducing costs, improving outcomes, and expanding access to care which will drive life-saving technologies to market. Thats honestly the reason were able to attract top talent from places like Google and Facebook. Especially in the data analytics world, I think the people who come here like the idea of working in a fun, productive culture, making a decent salary, getting stock that could be worth quite a bit down the road, and just generating the karmic return of doing something that truly is meaningful and impactful. James: I can definitely see TheraNova being a good role model and leading the younger industry by example. Im looking forward to working more together in the future. Dan: Thanks. Along those lines weve also started a nonprofit. Weve begun to assign rights to TheraNova technologies in Africa, India, and Southeast Asia to the nonprofit so that the technologies can be provided at lower costs to some of those areas. The nonprofits job is to adapt the technologies so they can be used in those much lower-cost areas. As an example of selecting the right investors and building the right board, Potrero Medical is, to my knowledge, the only venture-backed medical device company to assign these rights to the developing world after an equity financing with an institutional investor. This speaks volumes about the ethical alignment of the founders, investors and board members. James: Thats great, because I know one of the hurdles with nonprofits is that maybe the intention is good, but the execution is difficult. But if you have technologies that are already doing well in the U.S. market, perhaps theres a higher chance of success in the global market? Dan: Right, and if you start to generate the evidence-based medicine here, but adapt it so that it has fewer bells and whistles but is still very functional and can be provided at really low cost for these very low-resource settings, it could be very impactful. James: Is there anything else youd like to highlight? Dan: Well, I hope you dont mind me adding that WSGR has been a great partner for us. The firm helped open a lot of doors, and its been the counsel to all of my spin-offs since Polymorphics in 2000, which was the very first one at MedVenture Associates and, of course, is now called TheraNova. So Im very appreciative, and just like selecting investors, I would encourage founders to also consider their attorneys to be key, fundamental partners whom they should select wisely. While its easier to switch legal counsel than investors, attorneys can be equally valuable if you pick the right one. Top 10 Frequently Asked Questions Related to Human Factors and Medical Device Design By Russell J. Branaghan, Ph.D., President and Chief Scientist, Research Collective Advances in healthcare, medicine, and medical devices continue to improve the health and well-being of our society. Benefits of these technological advances include longer, healthier lives, quicker post-operative recovery times, and less time spent in hospitals. Despite these benefits, new technologies have the potential to introduce new and possibly unforeseen risks. For example, negative pressure wound therapy (NPWT) has been shown to reduce the healing time of wounds and burns. It promotes wound healing by applying a dressing and vacuum that not only draws fluid from the wound, but increases blood flow to the area. However, if the dressing is applied incorrectly, the vacuum can pull blood out of the wound, possibly leading to injury or death. In the case of NPWT, the only difference between improved healing and possible death is the way the dressing is applied by a human user. Careful consideration must be given to this interaction between the user and the technology. Human factors is the scientific study of how people interact with products, environments, systems, and services. Human factors can also be referred to as usability engineering, ergonomics, or user experience. For medical devices, human factors practitioners are responsible for ensuring that the design of the device, the instructions, and the training all support safe and effective use. (For a detailed description of human factors and its role in medical device design, see Shannon Clarks article in the Summer 2017 edition of The Life Sciences Report.) In recent years, the agencies that regulate medical devices have increased their demand for human factors to be considered in device design. In 2015, the International Electrotechnical Commission (IEC) released IEC 62366-1, Application of usability engineering to medical devices. A year later, the U.S. Food and Drug Administration (FDA) published its guidance document, Applying Human Factors and Usability Engineering to Medical Devices. These two documents describe the human factors activities manufacturers should apply when designing medical devices. Figure 1 provides a threestep approach to complying with IEC 62366-1 and the FDAs human factors guidance. As human factors consultants working with medical device manufacturers, we find that many manufacturers have similar questions regarding the human factors requirements. The following is a list of the top 10 most frequently asked questions (FAQs) and their answers.
1. How many use errors will the FDA accept? To quote the FDA directly, it depends. Its an unsatisfying answer, but a number of variables play a role:
Each use error, difficulty, or close call requires an in-depth analysis to determine the root cause. From there, the medical device manufacturer will need to decide whether the residual risk requires additional mitigation, or explain why the residual risk is acceptable as is. 2. Do we have to evaluate tasks that arent critical? If so, should noncritical task results be included in the report? The FDA is primarily interested in the results of critical tasks. However, evaluating every possible task is advisable. Evaluating noncritical tasks provides an opportunity to further understand the use of the medical device and to improve it. It is also prudent to have simulated-use data available for each task, should the FDA request it. Non-critical task results are not typically provided in detail. Instead, a summary of non-critical task results is appended to the report. This allows the reviewer a high-level view, with the ability to probe further if he/she deems it necessary. 3. How do we define critical tasks? The FDAs critical task definition is a good place to start. A critical task is a user task that, if performed incorrectly or not performed at all, would or could cause serious harm to the patient or user. In this case, harm is defined to include compromised medical care. Critical tasks should be determined from severity scores given in a risk analysis or failure modes and effects analysis (FMEA). While the risk analysis or FMEA traditionally gives each user task two scores—severity and probability—critical tasks are not concerned with probability. Any task that could lead to harm, regardless of likelihood of occurrence, should be listed as a critical task. User tasks related to successful delivery of therapy are often miscategorized as non-critical tasks. The FDA expects to see evidence that a device can be used for its intended purpose. Otherwise, what is the value of that device? 4. How realistic does the simulated-use environment need to be? The goal of a simulated-use usability study is to observe users interacting with the device interface independently and naturally. The FDA guidance recommends that testing take place in realistic but simulated use scenarios. There are three human factors considerations that interact to produce device use: the user, the device interface, and the use environment. Ultimately, the simulated-use environment should be realistic enough to understand how the device, the user, and use environment all interact. The design of simulated-use usability studies should consider all aspects of the use environment that may influence the users interaction with the device. Some of these aspects may include:
5. Can we make changes to the device or instructions after the validation usability study? Changes can be made after the validation study. However, it is likely the FDA would ask for a reevaluation of the tasks that were affected by the change. Only the tasks affected by the manufacturers change would need further evaluation. For example, if the wording of only one step in the instructions were changed, the FDA would likely ask for only that step to be reevaluated. Completing a reevaluation of only a few tasks is obviously a simpler and quicker affair, but it is important to note that a full 15 new participants would have to be included. Additionally, in order to reevaluate Step 4 of the instructions, participants would be asked to complete Steps 1-9 to ensure proper context. 6. What is the purpose of identifying known issues and how do we identify them? Known issues refer to usability issues that have already been documented from previous versions of the device, predicate devices, or devices that are similar. Recommending their identification is the FDAs way of preventing the same mistake from being made twice. By identifying usability issues with current and previous devices, manufacturers can avoid developing medical devices with the same problems. There are multiple places to look when searching for known issues related to usability, including, but not limited to:
7. What does the FDA mean by user groups for my device? Many medical devices are used by a diverse group of healthcare providers. For example, an injection device may be used by a nurse, physician, medical assistant, pharmacist, or a layperson with no training. User groups are simply these different roles within healthcare. In the case of an insulin pen, a doctor or nurse might demonstrate proper injection technique to their patient. The patient would then be responsible for performing his or her own injections thereafter. User groups for the insulin pen would be nurses, physicians, and patients (lay users). While performing an injection requires the same technique for each group, nurses, physicians, and lay users vary from one another in that they approach the task with different knowledge, experience, and training. 8. Can nurses and physicians be included in one user group? The FDA recommends that validation usability studies include 15 participants per user group. In an effort to save time and resources, medical device manufacturers often attempt to combine user groups. While there are situations where this may be pertinent, it is not typically appropriate. Despite working in similar environments, nurses and physicians are distinct from a human factors perspective. In addition to contrasting training and experience, nurses and physicians roles within healthcare vary greatly. Responsibilities tasked to a nurse do not typically align with those of a physician, and therefore the two are usually treated separately during validation usability testing. 9. How do you recommend that we incorporate user research into our design process? How often and when should we conduct user research? What are the best strategies? Incorporating human factors into the design process is most effective when done in the early stages of the product life cycle. Having the human factors perspective early on promotes informed decisions when the design is still flexible. Fundamental to early human factors success is the fact that late changes are difficult and expensive, and early ones arent. Early human-centered design is the most time-, effort-, and resource-efficient strategy. In 2017, the Association for the Advancement of Medical Instrumentation (AAMI) released a technical information report describing methods for incorporating human factors into design controls (see AAMI TIR59:2017 Integrating human factors into design controls). The key is iteration. Conduct user research to understand the users needs. Make design decisions with that information. Implement those designs into something testable. Lastly, have users interact with that prototype and get their thoughts on it. It doesnt matter how crude or developed it is; users will have something to say. Research, design, create, test, repeat. Many manufacturers find themselves severely confined by years-old engineering or design decisions that wouldnt exist if user research had been included early on. Investing in the human factors perspective earlier, rather than later, is strongly recommended. 10. Is there a fast and effective way to get feedback on the usability of my device without having to do an actual study with users? Usability studies are a powerful means of understanding the usability of a medical device. Accordingly, they tend to be the most time-consuming and expensive option. There are quicker, more cost-effective methods of evaluating usability. A heuristic analysis completed by a human factors expert compares a medical device to a set of design principles (often called heuristics). A heuristic analysis can be an extremely valuable, cost-effective tool to not only identify usability issues, but begin the process of making design improvements in the devices user interface. Conclusion The human factors discipline is increasingly recognized by the FDA, and is gaining traction within healthcare accordingly. While the FDAs human factors guidance document is intended to provide clarity on the topic, requests for further clarification persist. By understanding the intricacies of the recommended human factors processes, medical device manufacturers can pursue FDA submissions with confidence, efficiency, and an increased likelihood of success.
Focusing on human factors and usability testing, Research Collectives network of human factors experts and user experience professionals helps companies make their products easy to learn, efficient to work with, and desirable to use. Reps and Warranties Insurance Coverage in the Life Sciences Sector Part One – by Emily Maier, National Group Leader, M&A Insurance, Woodruff-Sawyer & Co. The life sciences M&A market shows no signs of slowing down. The total value of 2018 life sciences M&A should once again surpass $200 billion. The lull in biopharma deal-making last year and the passage of U.S. tax reform have sparked an increase in the desire and need for growth by acquisition. We are also witnessing the emergence of disruptive firepower in tech leaders Amazon, Alphabet, Apple, Microsoft, Intel, IBM, and Samsung, as well as retail giants CVS, Walgreens, and Walmart, all of which are openly moving into the life sciences and healthcare spaces for the first time. This is fueling M&A in these sectors as these disruptive players seek to purchase existing companies rather than organically develop the healthcare and/or life sciences expertise needed to execute their strategy. The fight for market share in key areas is affecting growth. We see an attack on pricing power as generic drug approvals surge in the U.S. and new products enter where pricing power had been relatively strong. The likely increased consolidation activity and new types of buyers will inevitably lead to increased use of representations and warranty insurance (RWI). As the head of the Woodruff Sawyer M&A Insurance Group, I have seen this already at play. RWI is breach-of-contract insurance. It covers the loss associated with discovering a breach of a rep or warranty after the deal has closed. Our experience is that corporate buyers use it more often when entering a new area. If we continue to see consolidation in a sellers environment, it will become a standard request by bidders. The RWI market has been cautious in life sciences, like most insurance carriers in this sector, because of phase III testing, the storage and usage of data, and the potential for very high payouts in the event of an issue. There are a couple of ways to minimize the risk of adding these exclusions to your reps policy. The first option is to make sure an exclusion clause is included in the RWI coverage (and I will offer some advice on that). The second form of protection is to understand what insurance coverages already are in place for the target. (My colleague Chad Follmer describes that in part two of this article.) These two strategies can and should go hand-in-hand. Products Liability In terms of risk, products liability is the principal insurable risk for most life sciences companies. In almost all reps and warranties policies, the coverage under the reps policy is designed to respond after any existing underlying insurances of the target have paid out. However, for life sciences companies, carriers may still seek to exclude products liability. Make sure that at the quote stage the broker is focusing on markets that are willing to provide coverage in excess of the existing target coverage. Cyber Liability This is a significant, ever-evolving concern for all industries, not just life sciences. It is particularly concerning for the life sciences industry because of the potential for large amounts of data protected by the Health Insurance Portability and Accountability Act (HIPAA) and the emerging bodily injury concerns posed by the risk of hacked medical devices. Harmonizing your products liability, medical professional liability, and cyber liability placements with your reps and warranties placement is of utmost importance to ensure you are getting the coverage you need. It is important that your underwriter understands exactly what the target does, and how data is used and stored. Dont assume that underwriters understand your business. They are specialists in reps and warranties insurance, not necessarily in life sciences. The more brokers involved who can help them understand the business, the better. We recently had an underwriter who insisted on a HIPAA exclusion. After we explained that the only testing done and the only records kept were for animals, they removed the exclusion. Part Two – by Chad Follmer, Healthcare Practice Leader, Woodruff-Sawyer & Co. The second way to protect yourself from financial harm in a purchase-sale transaction is to thoroughly understand the risk management measures taken and the insurance program of your target. This can be achieved through a proper risk management and insurance due-diligence analysis performed by a knowledgeable healthcare and life sciences specialist insurance broker. Its important to be sure that the target company has adequate limits and coverage breadth for the traditional areas of life sciences industry risk, such as products liability, clinical trials, privacy liability and network security, supply chain, and inventory risks, including spoilage. Questions to ask and areas to probe regarding these risks include the following:
While these traditional sector risks are important considerations, it is also critical to consider the evolving risks facing the life sciences industry, such as:
Finally, it is critical that your broker work in partnership with knowledgeable counsel to address the management liability risks associated with the deal itself, such as pricing, investor concerns, and disclosures, particularly for public companies. Add to this the unique regulatory concerns for the industry, including compliance with the Food and Drug Administration and the Centers for Medicare & Medicaid Services, and its clearly integral to have the right team of industry specialist brokers and attorneys on your side. Right now is a dynamic time in insuring the life sciences, with literally life-changing opportunity in our industry. The scale and impact of successful market adoption is greater than ever before, as are the risks. A well-executed risk management diligence audit in combination with a properly structured reps and warranties insurance placement are important tools for seizing the opportunities presented by strategic transactions. About the Authors
For 20 years, Chad has been bringing unique and proactive solutions to clients in the healthcare and life science industry. A leader in his field, Chad has a deep understanding of the complex risks facing these organizations, and knows how to create and deliver the optimal risk management solutions to protect their assets, people, and reputations. Chad specializes in the risks modern organizations in the healthcare and life science industry face, including: products and professional liability; regulatory risks; data privacy and cyber risks; alternative risk finance structures, such as captives, RRGs, SIRs, and trusts; and global risk management programs. Chad has led several practices in the area of risk management. He was most recently managing director, health care and life science practice leader for the western region of Marsh. Chad is a Risk and Insurance Power Broker award recipient and is a member of, and frequent presenter at, leading industry associations such as ASHRM, CAHF and HFMA. He authors the blog The Virtual Housecall: Healthcare Trends and Risks.
Leading Woodruff Sawyers M&A practice group, Emily provides consultation and support to clients who wish to use the insurance market to ring fence the risks that arise from M&A activity. This includes representations and warranties, tax opinion liability, and litigation buyout coverages. Emilys deal experience covers both American and European deals. Her clients are as diverse as their locations. She has worked with both strategic and private equity buyers and sellers over a wide range of transaction sizes and industries. Prior to joining Woodruff Sawyer in the U.S., Emily specialized in M&A insurance transactions in leading brokerage firms in London, including Marsh, Howden and Heath Lambert. Emily is a popular speaker and published author on the topic of M&A insurance. About Woodruff-Sawyer & Co. As one of the largest insurance brokerage and consulting firms in the U.S., Woodruff Sawyer protects the people and assets of more than 4,000 companies. Woodruff Sawyer provides expert counsel and advocacy to protect clients against their most critical risks in property and casualty, management liability, cyber liability, employee benefits, and personal wealth management. An active partner of Assurex Global and International Benefits Network, Woodruff Sawyer has headquarters in San Francisco, offices throughout the Life Sciences Venture Financings for WSGR Clients By Scott Murano, Partner (Palo Alto)
The data demonstrates that venture financing activity decreased from the second half of 2017 to the first half of 2018 with respect to the total amount raised and the total number of closings. Specifically, the total amount raised across all industry segments decreased 44.8 percent, from $1,792.4 million to $989.67 million, while the total number of closings across all industry segments decreased 14 percent, from 107 to 92. Notably, the industry segment with the largest number of closings—biopharmaceuticals—experienced a decrease in both number of closings and total amount raised from the second half of 2017 to the first half of 2018. Specifically, the number of closings in the biopharmaceuticals segment decreased 17.1 percent, from 41 to 34, and the total amount raised decreased 61.1 percent, from $1,062.65 million to $413.7 million. Similarly, the industry segment with the second-largest number of closingsmedical devices and equipmentexperienced a decrease in both number of closings and total amount raised from the second half of 2017 to the first half of 2018. Specifically, the number of closings in medical devices and equipment decreased 15.4 percent, from 39 to 33, and more significantly, the total amount raised decreased 33.8 percent, from $419.22 million to $277.61 million. Meanwhile, the industry segment with the third-largest number of closings during the second half of 2017—health IT—experienced decreases in both number of closings and total amount raised: the number of closings decreased 71.4 percent, from 14 to 4, while the total amount raised decreased 85.4 percent, from $184.45 million to $26.92 million. In contrast, the industry segment with the fourth-largest number of closings during the second half of 2017—healthcare services—experienced increases in both number of closings and total amount raised. Specifically, the number of closings in the healthcare services segment increased 57.1 percent, from 7 to 11, while the total amount raised increased 235 percent, from $53.7 million to $179.89 million. The two remaining industry segments—genomics and diagnostics—both experienced increases in number of closings from the second half of 2017 to the first half of 2018. The total amount raised in genomics increased 1,001.5 percent across those same periods, from $3.38 million to $37.23 million, while the total amount raised in diagnostics decreased 21.3 percent, from $69 million to $54.32 million. In addition, our data suggests that Series A (including Series Seed) financing activity and Series C and later-stage financing activity, in each case as a percentage of all other financing activity, increased from the second half of 2017 to the first half of 2018, while Series B financing activity as a percentage of all other financing activity, decreased across the same periods. Specifically, the number of Series A (including Series Seed) closings as a percentage of all closings increased from 35.2 percent to 36.6 percent, the number of Series C and later-stage closings as a percentage of all closings increased from 13 percent to 17.2 percent, and the number of Series B closings as a percentage of all closings decreased from 19.4 percent to 17.2 percent. Bridge financing activity as a percentage of all other financing activity decreased marginally from the second half of 2017 to the first half of 2018, moving from 23.1 percent to 22.6 percent. Average pre-money valuations for life sciences companies decreased for both Series A and Series B financings but increased for Series C and later-stage financings from the second half of 2017 to the first half of 2018. The average pre-money valuation for Series A financings decreased 56.8 percent, from $31.17 million to $13.45 million; the average pre-money valuation for Series B financings decreased 53.6 percent, from $124.14 million to $57.64 million; and the average pre-money valuation for Series C and later-stage financings increased 4.4 percent, from $225.91 million to $235.95 million. Other data taken from transactions in which all firm clients participated in the first half of 2018 suggests that life sciences remains the second-most attractive industry for investment. For the first half of 2018, life sciences represented 23 percent of total funds raised by our clients, while the software industry—traditionally the most popular industry for investment—represented 32 percent of total funds raised. Overall, the data indicates that access to venture capital for the life sciences industry decreased in the first half of 2018 compared to the second half of 2017, with the most active segmentsbiopharmaceuticals and medical devicesexperiencing significant drops in both number of closings and dollars raised. However, it is worth noting that this decline in activity follows several periods of consistent growth. Because recent exit activity for our life sciences companies remains strong across all industry segments, we are optimistic that the recent slowdown in financing activity represents nothing more than a natural cooling-off period after the recent growth, and should not serve as an indicator of things to come.
Select Recent Life Sciences Client Highlights
IRIDEX Announces Pricing of Public Offering of Common Stock
Phoenix 2018: The Medical Device and Diagnostic Conference for CEOs The 25th Annual Phoenix Conference will bring together top-level executives from large healthcare companies and CEOs of small, venture-backed firms for an opportunity to discuss critical issues of interest to the medical device industry today, as well as to network and gain valuable insights from both industry leaders and peers. This exclusive, two-day event will provide an unrivaled experience that will help inform and shape company strategy for the years ahead.
Bohemian Medical Device Summit This invitation-only conference for senior executives from medical device companies in Europe provides a private and exclusive setting for networking and partnering. The agenda will include high-level panel discussions with keynotes from industry and academic leaders, as well as social networking events. WSGR is one of the three original founding organizations of the Bohemian Medical Device Summit.
Women in Life Sciences Reception The Women in Life Sciences Reception will host women leaders in the life sciences industry for a lively evening of conversation and networking.
Biotech Board of Directors and Senior
Executives Reception Wilson Sonsini Goodrich & Rosatis annual Biotech Board of Directors and Senior Executives Reception, held to coincide with the J.P. Morgan 37th Annual Healthcare Conference, is an exclusive networking event geared toward executives and directors of biotechnology companies.
27th Annual Medical Device Conference Wilson Sonsini Goodrich and Rosatis 26th Annual Medical Device Conference will feature industry experts discussing key issues facing today's early-stage medical device companies. Through a series of topical panels, attendees will hear from industry CEOs, venture capitalists, industry strategists, investment bankers, and market analysts. The conference will kick off with a dinner on June 20.
Click here for a printable version of The Life Sciences Report This communication is provided as a service to our clients and friends and is for informational purposes only. It is not intended to create an attorney-client relationship or constitute an advertisement, a solicitation, or professional advice as to any particular situation. © 2018 Wilson Sonsini Goodrich & Rosati, Professional Corporation |
 Dan: Its about $300 million now that Potrero Medical just raised close to $27 million. What we do is, we take a technology and we do benchtop and preclinical work. Usually TheraNova pays for that. Then, if it passes muster, we move toward the clinical data. At any point along the way, if the technology fails, we scrap it.
Dan: Its about $300 million now that Potrero Medical just raised close to $27 million. What we do is, we take a technology and we do benchtop and preclinical work. Usually TheraNova pays for that. Then, if it passes muster, we move toward the clinical data. At any point along the way, if the technology fails, we scrap it.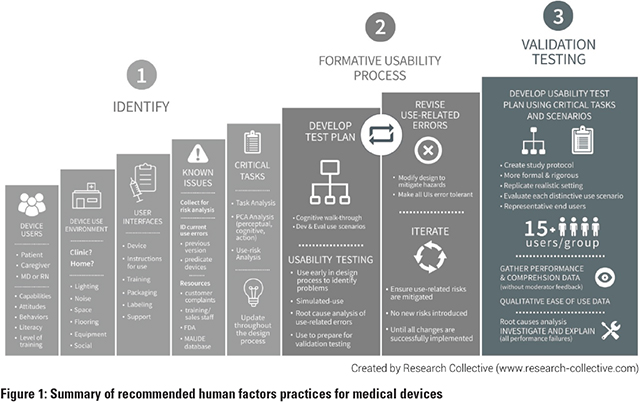
 Russell J. Branaghan, Ph.D. is
the president, chief scientist,
and founder of Research
Collective, a usability testing
lab and consultancy in Tempe,
Arizona. Russ is a professor
of cognitive psychology and human factors
at Arizona State University and a longstanding
visiting professor at Northwestern
University. He is integrated in the human
factors arena, where he currently serves as
chair of the Human Factors and Ergonomics
Society Product Design Technical Group, on
the editorial board of the Journal of Human
Factors, and on the advisory board for
Mayo Clinics Center for Innovation. An avid
researcher and writer, Russ has authored over
40 articles and one book. He can be reached
at
Russell J. Branaghan, Ph.D. is
the president, chief scientist,
and founder of Research
Collective, a usability testing
lab and consultancy in Tempe,
Arizona. Russ is a professor
of cognitive psychology and human factors
at Arizona State University and a longstanding
visiting professor at Northwestern
University. He is integrated in the human
factors arena, where he currently serves as
chair of the Human Factors and Ergonomics
Society Product Design Technical Group, on
the editorial board of the Journal of Human
Factors, and on the advisory board for
Mayo Clinics Center for Innovation. An avid
researcher and writer, Russ has authored over
40 articles and one book. He can be reached
at  Chad Follmer, Healthcare Practice Leader, Woodruff-Sawyer & Co.
Chad Follmer, Healthcare Practice Leader, Woodruff-Sawyer & Co. Emily Maier, National Group Leader, M&A Insurance, Woodruff-Sawyer & Co.
Emily Maier, National Group Leader, M&A Insurance, Woodruff-Sawyer & Co.




