
Spring 2013 Best Patent Practices Under the America Invents Act By Charles Andres, Associate (Washington, D.C.), Esther Kepplinger, Chief Patent Counselor (Washington, D.C., and San Diego), Jasemine Chambers, Of Counsel (Washington, D.C.), and Vern Norviel, Partner (Palo Alto and San Diego) On March 16, 2013, the first-inventor-to-file provisions of the America Invents Act (AIA) took effect. These provisions, which apply to applications with an effective filing date on or after March 16, 2013, fundamentally alter United States patent law that has been in effect for the last 60-plus years. For example, under the AIA, patents will be awarded to the first inventor to file a patent application, not the first inventor to invent. Also, the universe of prior art that can be applied against a patent application is substantially enlarged. Further, pending patent applications and issued patents may now be challenged by a variety of U.S. Patent and Trademark Office procedures, and these challenges can be resolved within relatively short periods of time. The challenge procedures are in addition to more traditional, lengthy, and expensive federal district and appellate court litigations. Because the AIA fundamentally altered the patent landscape, companies that evolve to function optimally in the new environment will enjoy a competitive advantage (i.e., survival of the fittest). Below we offer 10 best practices that will be generally applicable and beneficial for patent applicants and patent owners. Best Practice No. 1: Promptly Draft and File Patent Applications Under the AIA, patents will generally be awarded to the first inventor to file a patent application. Companies that streamline the process from invention disclosure to application filing will increase their odds of filing first and give themselves an advantage over their competitors. Best Practice No. 2: Use Provisional Patent Applications to Maximize Advantage Because the AIA rewards first filing, consider filing provisional patent applications on all inventions first and deciding on the significance of the inventions later (budget permitting). Also, consider filing a follow-on provisional application each time a new improvement is realized. Best Practice No. 3: Budget Now for Increased Patent Application Drafting, Filing, and Prosecution Costs The AIA may necessitate drafting and filing more provisional patent applications, and filing and prosecuting more non-provisional patent applications. Consider increasing your intellectual property (IP) budget now to deal with these eventualities. Best Practice No. 4: Consider Filing Patent Applications Under Your Company Name It is now possible to file U.S. patent applications in the name of the assignee (e.g., the company). Thus, companies should consider filing patent applications in their own name, as this offers several legal advantages under the AIA. Best Practice No. 5: File Provisional Applications Before Any Public Disclosure Publically disclosing an invention before filing a patent application is best avoided. Pre-application-filing public disclosure almost always destroys the ability to patent an invention outside of the United States. The AIA provides limited exceptions to the general rule in the U.S., but it is best not to rely on the limited exceptions. Consider filing provisional applications before allowing any public disclosure of an invention. Best Practice No. 6: Obtain Joint Research Agreements Now If a joint research agreement is in place when (or before) an application is effectively filed, the AIA allows for the removal of some art that would otherwise be prior art. Is your company collaborating with a university or another company? Consider putting joint research agreements in place now. Best Practice No. 7: Interview Cases with Patent Examiners Examiner-interviewed patent applications are allowed faster and more often than applications prosecuted without examiner interviews. Also, recent court decisions make interviewing certain cases a best practice. Interviewing can shorten prosecution time, lower costs, minimize estoppel, and maximize claim scope. Consider interviewing some or all of your cases (budget permitting). Best Practice No. 8: Be Prepared to Have Patent Applications and Issued Patents Challenged The AIA provides a variety of mechanisms for competitors to challenge a patent after it is granted. Also provided is a new mechanism for competitors to submit art and comments during the prosecution of a patent application. Consider preparing for such challenges by taking preemptive actions, such as including claims of different scopes, including "fall back" positions when drafting an application, and maintaining an awareness of the art in your technology space. Best Practice No. 9: Monitor and—When Appropriate—Challenge Competitor Applications and Patents The AIA provides mechanisms to challenge granted patents and to submit art and comments during the prosecution of a patent application. A well-crafted, adequately supported challenge can force a competitor to limit claim scope and open up freedom to operate. A timely challenge can delay or destroy a pending competitor deal (e.g., for funding or sale). Consider monitoring competitor patent portfolios and selectively using challenges to shape the behavior of competitors in a manner favorable to your company. Best Practice No. 10: Address the Need for Laboratory Notebooks and How Experiments Are Recorded and Maintained Now that there is no need to maintain laboratory notebooks for patent interference reasons, a primary patent-related legal reason to maintain such notebooks is to prove inventorship. Companies should therefore seriously consider addressing the need for laboratory notebooks in the future. Considerations that might cause a company to continue maintenance of laboratory notebooks include: a) the need for such records for FDA purposes, b) the desire of the scientist to maintain records of his experiments in a "familiar" manner, or c) the need to prove inventorship in the unlikely case where computer records are not adequate. Considerations that might lead companies to discontinue the requirement for scientists to maintain laboratory notebooks include: a) the cost and time required to maintain notebooks, b) the availability of complete electronic records, and c) the potential for material in the notebooks to be used against a company in later patent litigation. Conclusion Companies that evolve their IP strategies to function optimally in the new AIA legal environment will enjoy a competitive advantage. Wilson Sonsini Goodrich & Rosati has the experience and know-how to help companies with this evolution. For assistance or more information, please contact any member of the firm's intellectual property practice.
CMS Issues Final Rule for Implementing Sunshine Act By David Hoffmeister, Partner, and Jon Nygaard, Attorney (Palo Alto) On February 8, 2013, 16 months after the statutory deadline, the Centers for Medicare & Medicaid Services (CMS) published in the Federal Register the final regulation implementing the physician payment transparency provisions—collectively known as the Physician Payment Sunshine Act—of the Patient Protection and Affordable Care Act of 2010 (the ACA). The rule became effective on April 9, 2013, 60 days after the publication date. Drug and device manufacturers will be required to track payments or transfers of value to physicians and teaching hospitals, and physician ownership and investment interests in such manufacturers, beginning on August 1, 2013, and their first report of such payments and ownership and investment interests (covering the last five months of 2013) will be due on March 31, 2014. Companies would be well advised to begin implementing systems, practices, and procedures that will allow them to begin tracking all relevant transactions and ownership and investment interests, if they have not started already. Transparency will impose a major burden on manufacturers, as compliance with the rule will exact from them considerable time and resources. Executive Summary The ACA requires manufacturers to submit two separate but related reports to the Department of Health and Human Services (HHS). First, applicable manufacturers of drugs, devices, biologicals, or medical supplies that are available for coverage under Medicare, Medicaid, or the Children's Health Insurance program (CHIP) must report annually to HHS certain payments or other transfers of value to1 covered recipients, namely, physicians and teaching hospitals. The rule provides definitions of numerous terms, such as "applicable manufacturer" and "covered drug, device, biological, or medical supply." In addition, it clarifies how applicable manufacturers should report and characterize payments or other transfers of value, including rules for research payments and indirect payments provided to a covered recipient through a third party. The rule also finalizes which payments are excluded from the reporting requirements. Second, applicable manufacturers and group purchasing organizations (GPOs) must report information on ownership and investment interests in such entities held by physicians or their immediate family members. The rule details what constitutes an ownership or investment interest and defines for whom they must be reported. The rule also clarifies the content of the report concerning ownership of investment interest. The rule finalizes the processes and requirements for applicable manufacturers and GPOs to submit their reports to CMS, including the specific data elements that are required to be included in the reports and the report format. It also details the processes for the review, dispute, and correction period when applicable manufacturers and GPOs, covered recipients, and physician owners or investors are provided the opportunity to review, dispute, and propose corrections to the reported payments, or ownership or investment interests, attributed to them. The rule clarifies the information to be included on the publicly available website, as well as the usability of the public website. In addition, the rule includes details on the processes for reporting and publishing payments that are eligible for delayed publication. Finally, the rule includes details regarding the statutorily authorized civil monetary penalties for failure to report payments, or physician ownership or investment interests. It also clarifies the statutory requirements for the preemption of state laws. Notable Changes to the Proposed Regulation Among the more significant changes to the proposed regulation issued by CMS in December 2011 are the following:
Nonetheless, CMS does not have much to show for the 16 months it took to finalize the proposed regulation issued in December 2011. One wonders whether CMS will have sufficient resources to monitor thousands of manufacturers for compliance with the ACA and final regulation. The remainder of this article summarizes some of the key provisions of the final rule. Reports on Payments and Other Transfers of Value (Transparency Reports) Who Must Submit Reports? Transparency reports must be submitted by "applicable manufacturers," which CMS defines as entities operating in the United States and falling into one of these two categories:
What Is a Covered Drug, Device, Biological, or Medical Supply? Any drug, device, biological, or medical supply for which "payment is available" under Medicare, Medicaid, or CHIP, either separately or as part of a bundled payment, is covered by the rule. Covered drugs or biologicals include only those that require a prescription, so over-the-counter (OTC) drugs are not covered. Devices or medical supplies are covered only if they require premarket approval by the U.S. Food and Drug Administration (FDA) or premarket notification (i.e., 510(k) clearance), so 510(k) Class I-exempt devices and 510(k) Class II-exempt devices are not covered. A product that is not approved or cleared nevertheless may be a covered product if "payment is available" for it under Medicare or Medicaid. For example, payment is available under Medicare for certain devices covered under an investigational device exemption (IDE), and payment may be available under Medicaid for certain unapproved pre-1962 prescription drugs. When a manufacturer's first product becomes eligible for payment under Medicare, Medicaid, or CHIP, CMS will allow a grace period of 180 days after the product becomes "covered" before the manufacturer must begin complying with the data collection and reporting requirements. Who Are Covered Recipients and How Are They Identified? "Covered recipients" are either physicians (other than bona fide employees of a manufacturer) or teaching hospitals. CMS defines "physicians" as doctors of medicine and osteopathy, dentists, podiatrists, optometrists, and chiropractors who are licensed by the state in which they practice. Other provider types, such as nurse practitioners or residents, are not included in the definition. Manufacturers must report a physician recipient's name, business address, National Provider Identifier (NPI), and specialty. If the physician's NPI is not available on the National Plan and Provider Enumeration System (NPPES) website, a manufacturer must make a good-faith effort to obtain the NPI from the physician; a "good-faith effort" is requesting an NPI from a physician, checking the NPPES database, and calling the NPPES help desk. If a manufacturer cannot determine the physician's NPI or the physician does not have one, the space may be left blank. CMS defines "teaching hospitals" as any institutions that received Graduate Medical Education payments under Medicare in the most recent year for which information is available. CMS will publish a list of all such hospitals annually, and manufacturers may rely on the list for the entire reporting year. What Payments or Transfers of Value Must Be Reported? The regulation requires manufacturers to report "direct and indirect payments or other transfers of value" to a covered recipient, or to a third party at the request of a covered recipient. It defines a "payment or other transfer of value" as "a transfer of anything of value." In determining reported value, CMS considers "value" to mean the discernible economic value on the open market in the U.S. All aspects of the value, such as taxes or shipping, should be included in the reported value. Beyond this, CMS declines to provide rules for calculating value. Manufacturers must make a reasonable, good-faith effort to determine the value of a payment or transfer of value, and may include the methodology used in a voluntary assumptions document that is submitted to CMS. A payment made "at the request of" a covered recipient means that the covered recipient has directed the manufacturer to provide the payment to another entity or individual rather than receiving it personally—for example, a fee waived by a physician and then donated by a manufacturer to a charity on behalf of the physician. Such payments are to be reported under the name of the covered recipient, but the report also should include the name of the entity that received the payment, or, if an individual received it, the designation "individual" (so as to preserve the privacy of such individuals). The rule sets forth a number of exclusions, which are described later in this article. What Are the Contents of the Report? Manufacturers must report to CMS the following information regarding any payment to a covered recipient:
The rule elaborates further on the "nature" of payments to be reported in general and for certain of the above categories: Payments with multiple categories: Only one nature may be indicated for each payment. If a payment could fit within several categories, the manufacturer should select the most suitable one, but should not bundle payments belonging to separate categories into a single payment. For example, a meal should be reported as a meal, even if it is associated with travel or a consulting contract. Charitable contributions: This category should only be used where an applicable manufacturer makes a payment to a charity on behalf of a covered recipient, but not in exchange for any service or benefit. For example, if a physician requests that his or her consulting fee be paid to a charity, this should be reported not as a charitable contribution, but as a consulting fee with the physician as the covered recipient and the charity as the entity paid. Meals and beverages: The cost of meals provided in a group setting must be divided by the total number of individuals who ate the meal (both physicians and non-physicians, such as office staff), with the resulting per-person cost reported for each physician who actually participated in the meal. Additionally, CMS is excluding the reporting of buffet meals, snacks, soft drinks, or coffee made generally available to all participants at large-scale conferences or similar events. Payments for CME and speaker fees: Payments for CME that is accredited by the ACCME or other specified accrediting organizations are exempt from reporting if the manufacturer does not pay faculty directly and does not select or recommend individual faculty members. If a CME program is accredited, but the manufacturer directly pays or recommends the faculty, the payments must be reported as "Compensation for serving as faculty or as a speaker for an accredited or certified continuing education program." If a program is not accredited, the payment is reported as "Compensation for serving as faculty or as a speaker for an unaccredited and non-certified continuing education program." Finally, where a payment is made to a physician speaker at an event that is not continuing medical education (for example, a promotional speaker program), the payment should be reported as "Compensation for services other than consulting, including serving as faculty or as a speaker at other than a continuing education program." "Other" Category Deleted: The final rule omits the "other" nature category in the proposed rule because it would dilute the usefulness of the "nature" categories. CMS cautions that all payments to covered recipients must be reported, and failure to identify a nature category could result in penalties. Therefore, manufacturers should select the nature category that most closely describes the payment. Research Payments Under the final rule, payments for research are reported separately from other payments and transfers of value, using a different reporting format. "Research" includes basic and applied research, preclinical research, Phase I through IV studies, and investigator-initiated studies. If a payment falls within the definition of "research" and is subject to a written agreement, a protocol, or both, it is reported as research. Research-related payments that do not meet these requirements must be reported using other "nature" categories. Manufacturers will not be required to attribute the entire research payment made to a facility to each principal investigator. Instead, the manufacturer will report each research payment once, identifying the name and address of the institution (whether or not a teaching hospital) or individual physician paid, the amount, the name of the study, the name of the related product, and information about each principal investigator. At their option, manufacturers may report explanatory information about the study. The requirements for reporting payments for pre-clinical studies are similar, but no associated product or study name need be reported. The ACA requires CMS to delay publication of payments from manufacturers to covered recipients made (1) pursuant to a product research or development agreement or (2) in connection with a clinical investigation regarding a covered product. Delayed publication will apply to payments for both research and development, and clinical investigations, where they relate to a new product. However, where a new application of an existing product is concerned, publication will be delayed for a research and development payment but not for a clinical investigation payment. In other words, payments to clinical investigators will be entitled to delayed publication if the investigation is for a new product, but not if it is for a new indication of a currently marketed product. The only payments for "research" of new indications of marketed products that would be subject to delayed disclosure would be payments for non-clinical studies. CMS clarifies that products for which approval or clearance will be sought under an Abbreviated New Drug Application (ANDA) or a 510(k) notification are considered new products, rather than new applications of existing products. Despite comments that CMS should not publish the payments until after FDA approval, licensure, or clearance, CMS stated in the preamble to the final rule that it believes "Congress clearly intended that all payments should be included on the public website, even if a product never received FDA approval, licensure or clearance." For publication to be delayed, the manufacturer must indicate in its transparency report whether a payment is eligible for a delay in publication. The failure to indicate eligibility will result in the payment being posted publicly in the following year. The manufacturer also must continue to indicate annually that FDA approval, license, or clearance is pending, and subsequently must notify CMS if the FDA approves or clears the product. Exclusions Under the statute and the final regulation, the following payments are excluded from the reporting requirements: (1) Transfer through a third party. No reporting is required for a transfer of value made indirectly to a covered recipient through a third party in cases where the applicable manufacturer does not know the identity of the covered recipient. Awareness of the identity of a recipient on the part of a legal agent acting on behalf of the manufacturer is attributed to the manufacturer itself. (2) De minimis payments. Payments and transfers of value less than $10 are not reportable, unless the aggregate amount transferred to, requested by, or designated on behalf of a covered recipient exceeds $100 in a calendar year. Small items that are under $10 (such as pens and notepads) that are provided at large-scale conferences and similar large-scale events are exempted from the reporting requirements, and also do not need to be tracked for purposes of the $100 aggregate threshold. (3) Samples. Product samples that are not intended to be sold, yet intended for patient use, including coupons and vouchers, are not reportable. (4) Educational materials. Educational materials that directly benefit patients or are intended to be used by or with patients are not reportable. CMS has clarified that this exemption does not cover materials provided to physicians for their own education, nor does it cover marketing or promotional materials. This narrow interpretation imposes a considerable burden on manufacturers, which must report the value of all reprints and promotional materials provided to physicians if they exceed the de minimis threshold throughout the year. (5) Devices for evaluation. Manufacturers need not report the loan of a covered device for a short-term trial period, not to exceed 90 days, or the provision of a limited quantity (i.e., 90 days of average use) of disposable or single-use devices or medical supplies, to permit evaluation of the covered device by the covered recipient. The exemption for disposable or single-use devices or supplies was added in the final rule. (6) Warranty items. Items or services provided under a contractual warranty (including a service or maintenance agreement), including the replacement of a covered device, are not reportable where the terms of the warranty are set forth in the purchase or lease agreement for the covered device. The exemption applies even if the warranty period has expired. (7) Charity care. Manufacturers are not required to report in-kind items used for the provision of charity care, defined as care for a patient who is unable to pay or for whom payment would be a significant hardship, where the covered recipient does not receive or expect to receive payment. This exemption does not include in-kind items provided to a charitable organization for the care of all of its patients, both those who can and cannot pay. Moreover, the exemption covers only in-kind items, not financial support for charity care. (8) Covered recipient who is a patient. Reporting is not required for a payment or transfer of value to a physician who is a patient, research subject, or participant in data collection for research, and not acting in the professional capacity of a physician. (9) Discounts and rebates are not reportable. (10) Publicly traded securities. A dividend or other profit distribution from, or ownership or investment interest in, a publicly traded security or mutual fund is not reportable. (11) Health care for employee. In the case of a manufacturer that offers a self-insured plan, payments for the provision of health care to employees under the plan are not reportable. (12) Payments for non-medical services. Where a physician is also a licensed non-medical professional, a payment to the physician is non-reportable if it is solely for the non-medical professional services of the individual. (13) Payments for services in judicial proceeding. Manufacturers need not report a payment to a physician if the payment is solely for the services of the physician with respect to a civil or criminal action or an administrative proceeding. (14) Personal relationship. A payment to a physician is not reportable if it is made solely in the context of a personal, non-business-related relationship. Reports on Physician Ownership and Investment Interests In addition to transparency reports, the ACA requires manufacturers and GPOs to separately report information on ownership or investment interests in such entities held by physicians or their immediate family members. CMS finalized its proposed definition of "applicable GPO" as an entity that operates in the United States and purchases, arranges for, or negotiates the purchase of a covered drug, device, biological, or medical supply for a group of individuals or entities, but not solely for use by the entity itself. CMS states that this definition includes purchasers and physician-owned distributors of covered drugs, devices, biologicals, and medical supplies, but does not include bulk purchasers for commonly owned entities. An "immediate family member" is defined as one of the following:
An ownership or investment interest in a manufacturer or GPO may include stock, stock options (when exercised), partnership shares, loans, and bonds. However, an ownership or investment interest does not include any publicly traded security or mutual fund. Manufacturers and GPOs need not report indirect ownership or investment interests held by physicians or their immediate family members about which the manufacturers or GPOs did not know. Manufacturers and GPOs must report the following information for each physician ownership or investment interest:
To avoid duplicative reporting, manufacturers should report the payments provided to physician owners or investors in the report for payments, and should note that the covered recipient receiving the payment or other transfers of value is a physician owner or investor. Additionally, an individual may be both a covered recipient and a physician owner or investor. A manufacturer should only report a payment once, regardless of whether it is required to be reported as a payment or an ownership interest. Report Submission and Correction Manufacturers and GPOs must submit their reports for the preceding calendar year electronically to CMS by March 31, 2014, and by the 90th day of each calendar year thereafter. Only manufacturers that made a payment to a covered recipient or had a physician owner or investor in the previous calendar year need to register and submit a report to CMS. Similarly, only GPOs with a physician owner or investor are required to submit a report. In other words, even if an entity meets the definition of "applicable manufacturer" or "applicable GPO," it need not register or submit a report if it has no payments or other transfers of value to report. A manufacturer under common ownership with separate entities that are also applicable manufacturers may, but is not required to, file a consolidated report of all payments, and physician ownership or investment interests, for all entities. All manufacturers with payments to report must register individually, even if they intend to be part of a consolidated report submitted by another manufacturer. Manufacturers submitting data as part of a consolidated report that will be submitted by another manufacturer may indicate during registration that they intend to be part of the report submitted by another manufacturer. The entity submitting the consolidated report must indicate all of the manufacturers for which it is reporting. An authorized representative must submit a signed attestation at the time of data submission certifying the truthfulness, accuracy, and completeness of the data submitted to the best of the signer's knowledge and belief. An entity submitting a consolidated report must attest on behalf of itself and each of the other manufacturers included in the report. While the attestation must be provided at the time of data submission, it also must be provided any time the data is changed or updated. Data without an attestation will not be considered an official submission. For a manufacturer with payments or other transfers of value to report, if covered products represent less than 10 percent of total (gross) revenue for the preceding year, the attestation must indicate that fact. CMS will not grant submission extensions, and any late data will be considered a failure to report, which may be subject to penalties. The statute requires that, following submission of the reports, CMS must provide manufacturers, GPOs, covered recipients, and physician owners and investors with the opportunity to review the data for at least 45 days prior to publication on the public website. If a covered recipient or physician owner or investor disagrees with the data, he can initiate a dispute, and applicable manufacturers or GPOs may begin resolving the dispute and correcting the data. After the end of the 45-day review-and-correction period, manufacturers and GPOs will have an additional 15 days to correct data for the purposes of resolving disputes, after which they may submit, and provide attestation for, the updated data to CMS to finalize the submission. Payments or ownership or investment interests that cannot be resolved by the end of the 15-day resolution period will be marked as "disputed," but the manufacturer's or GPO's most recent attested data subject to the dispute will be the only information published. The 45-day review-and-correction period and 15-day dispute-resolution period will not be the only opportunities to dispute the contents of the public website. CMS will allow physicians and teaching hospitals, and physician owners and investors, the opportunity to sign in to the system to review or dispute officially submitted and attested transactions any time during the year. Any disputes resolved outside the 45- and 15-day time periods, however, will not be reflected on the public website until the next update of the data. Civil Monetary Penalties If a manufacturer or GPO fails to submit the required information, it may be subject to penalties of not less than $1,000, but not more than $10,000, for each payment or ownership or investment interest not reported. The maximum penalty that can be assessed for failure to report is $150,000 each year. For knowing failures, a manufacturer or GPO will be subject to penalties of not less than $10,000, but not more than $100,000, for each payment or ownership or investment interest not reported. The maximum penalty for a knowing failure to report is $1,000,000 each year. The penalties imposed on each manufacturer or GPO are aggregated separately, and subject to separate aggregate totals for failures to report and knowing failures to report, with a maximum combined total of $1,150,000. The factors that CMS will consider in determining the amount of a penalty include, but are not limited to, the following:
For consolidated reports, the manufacturer that submits the consolidated report will be required to attest on behalf of all the entities included in the consolidated report, and therefore will be subject to the maximum penalties for each individual manufacturer included in the report. The submitter of the consolidated report therefore could be subject to a penalty greater than $1,000,000 depending on the violations of the manufacturers for whom it submitted the report and attested as to the data. HHS, CMS, the Office of the Inspector General (OIG), or their designees have the right to audit or inspect manufacturers or GPOs to assess their compliance with the requirement to provide timely, complete, and accurate submissions of the information. In order to facilitate the auditing and inspection process, manufacturers and GPOs must maintain books, records, and documents to enable an audit or inspection for a period of at least five years from the date the payment or other transfer of value, or ownership or investment interest, is published on the website. CMS Website The statute requires CMS to publish the data collected from manufacturers and GPOs on a publicly available website by June 30 of each year. Due to the timing of the final rule, the first publication will be in June 2014 for data collected in 2013. In the preamble to the final rule, CMS stated that it plans to engage stakeholders regarding the content of the website, since it recognizes that stakeholders and the public must be part of the website-development process. CMS will ensure that the website "accurately and completely describes the nature of relationships between physicians and teaching hospitals, and the industry, including an explanation of beneficial interactions," and that it will clearly state that disclosure on the website "does not indicate that the payment was legitimate nor does it necessarily indicate a conflict of interest or any wrongdoing." Annual Reports CMS must submit annual reports to Congress and the states. The annual report is due to Congress on April 1 of each year, and must include aggregated information on each manufacturer and GPO submitted during the prior year, as well as any enforcement actions taken and penalties paid. The state reports will be state-specific. Relation to State Laws The ACA preempts any state or local laws requiring the reporting of the same type of information regarding payments made by applicable manufacturers to covered recipients. However, this does not prevent a state from collecting this information for public-health surveillance, investigation, or other public-health purposes or health oversight. The public-health goal must be one other than transparency in order for the state reporting to avoid preemption. State and local governments may require the reporting of information other than that required under the ACA. The additional information may include other types of information (except payments that fall below the $10 individual or $100 aggregate thresholds), or payments to health care providers other than physicians and teaching hospitals. Conclusion Manufacturers and GPOs must begin tracking payments to physicians and teaching hospitals, as well as physician ownership and investment interests, starting on August 1, 2013, in order to report them to CMS by March 31, 2014. They would be well advised to implement systems, practices, and procedures to accomplish these tasks in the two or so months remaining.
1 Generally, this article will refer to "payments or transfers of value" simply as "payments." New IP Litigation Team a Boon for Firm's Life Sciences and Technology Clients Wilson Sonsini Goodrich & Rosati Adds Five New Partners—and an LA Office
Life Sciences Venture Financings for WSGR Clients By Scott Murano, Partner (Palo Alto)
The data generally demonstrates that venture financing activity declined during the second half of 2012 compared to the first half of 2012. Specifically, the total number of closings completed across all industry segments during the second half of 2012 decreased by 9 percent compared to the first half of 2012, from 111 closings to 101 closings. More significantly, the total amount of money raised across all industry segments during the second half of 2012 decreased by more than 20.1 percent compared to the first half of 2012, from $619.18 million to $494.49 million. The three industry segments with the largest number of closings—medical devices and equipment, biopharmaceuticals, and diagnostics—were the only segments to experience a decline in number of closings during the second half of 2012 compared to the first half. Specifically, the largest industry segment, medical devices and equipment, declined 9.6 percent, from 73 closings in the first half of 2012 to 66 closings in the second half; the second-largest industry segment, biopharmaceuticals, declined 13.3 percent, from 15 closings to 13 closings; and the third-largest industry segment, diagnostics, declined 40 percent, from 10 closings to 6 closings. In terms of total amounts raised, all identified industry segments other than genomics experienced a decline in the second half of 2012 compared to the first half. Most notably, healthcare services declined 77.4 percent, from $35.04 million during the first half to $7.93 million during the second half, and medical information systems declined 67.1 percent, from $33.56 million during the first half to $11.04 million during the second half—and in both cases the industry segment had the same number of closings in both halves of 2012. The experience of these industry segments, along with the industry generally, demonstrates that the decline in the total amount raised is disproportionately outpacing the decline in number of closings, which may suggest that investors are expecting companies to do more with less capital. In addition, our data suggests that Series A financing activity is up compared to Series B and later-stage equity financings and bridge financings. Specifically, the number of Series A closings as a percentage of all closings during the second half of 2012 compared to the first half of 2012 increased from 24.3 percent to 28.7 percent, whereas the number of Series B closings during the same periods decreased from 17.1 percent to 15.8 percent, the number of Series C and later closings decreased from 19.8 percent to 15.8 percent, and the number of bridge financings decreased from 37.8 percent to 35.6 percent. Moreover, the data demonstrates that the average pre-money valuations for Series A and Series B closings declined, while the average pre-money valuations for Series C and later closings increased. Specifically, the average pre-money valuation for Series A financings fell 47.4 percent, from $13.64 million during the first half of 2012 to $7.17 million during the second half; Series B financings fell 6.3 percent, from $18.61 million to $17.43 million; and Series C financings increased by 28.2 percent, from $106.34 million to $136.35 million. The drop in average Series A pre-money valuations from the first half of 2012 may help to explain the increase in Series A activity relative to later-stage equity or bridge investments during the second half of 2012. Other data taken from transactions in which all Wilson Sonsini Goodrich & Rosati clients participated in 2012 suggests a shift in investment money from life sciences to other industries. In the first half of 2012, life sciences was the second-most attractive industry for investment among our clients, representing 20.1 percent of total funds raised, and was barely edged out of the number one spot by the software industry, which represented 20.4 percent of total funds raised. Clean technology and renewable energy followed in third place with a distant 14.6 percent of total funds raised. In the second half of 2012, life sciences retained the number two spot at 19.2 percent of total funds raised, with software gaining more ground on life sciences as the top industry for investment at 24.9 percent, and clean technology and renewable energy remaining in the third spot but gaining ground on life sciences, at 18.1 percent. Overall, the data confirms that access to venture capital for life science companies declined during the second half of 2012 compared to the first half of 2012, and it is important to note that I last reported in our Fall 2012 Life Sciences Report that venture activity during the first half of 2012 had declined from the second half of 2011. While life sciences once stood as the highest-grossing industry for investment among our clients, it continues to lose ground to software as the number one industry for investment, and may at some point be surpassed by clean technology and renewable energy. Critics of the life science industry point to regulatory uncertainty, a contraction of venture capital firms, longer exit times, poor returns, and a more limited appetite among would-be acquirers for acquisitions. The upshot in the data may just be that Series A activity for life science companies was up marginally during the second half of 2012 compared to the first half, not just as a relative percentage of closings at all stages of investment, but in terms of the absolute number of Series A closings, suggesting that there remains interest in early-stage investment, but at lower pre-money valuations and with less dollars raised over time—potentially signs of an industry redefining itself and its priorities.
Strategies for Maximizing Patent Claim Scope and Patent Protection for Diagnostic Method Claims in the Wake of Mayo v. Prometheus By Charles Andres, Associate (Washington, D.C.), Vern Norviel, Partner (Palo Alto and San Diego), Esther Kepplinger, Chief Patent Counselor (Washington, D.C., and San Diego), Jasemine Chambers, Of Counsel (Washington, D.C.), Louis Lieto, Associate (Washington, D.C.), and Christopher McAndrew, Patent Agent (Washington, D.C.) Background The United States Supreme Court recently sent a shockwave through the biotechnology industry by issuing an important decision on patent claims drawn to correlative diagnostic methods1 practiced in the area of personalized medicine.2 In Mayo v. Prometheus,3 the Supreme Court assessed whether claims in U.S. Patent Nos. 6,680,302 and 6,355,623 were patent eligible.4 Representative claim 1 of the '623 patent5 was directed to a process for individually optimizing the dosage of 6-thioguanine for treatment of an immune-mediated gastrointestinal disorder. 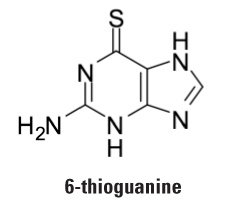 6-Thioguanine is used for the treatment of human inflammatory bowel disease, ulcerative colitis, and celiac disease, all of which are autoimmune-mediated gastrointestinal disorders.6 To optimize therapeutic effectiveness, 6-thioguanine (drug) must be administered at a dose that falls within a therapeutic window.7 If the dose is too low, the drug will not work. If the dose is too high, unwanted side effects may emerge. Because individual patients will metabolize 6-thioguanine differently, "personalized" dosages of 6-thioguanine may be required for each patient. Claim 1 of the '623 patent was drawn to a method of optimizing therapeutic efficacy (dose) of 6-thioguanine for treatment of an immune-mediated gastrointestinal disorder. Claim 1 required:
Claim 1 also recited concentration correlations (in wherein clauses) suggesting when subsequent dosages of 6-thioguanine should be increased or decreased, but did not actually require that subsequent dosages of 6-thioguanine be adjusted if the correlations were met. The Court viewed these correlations as laws of nature.8 The Supreme Court in Prometheus declared claim 1 (and the related claims) to be patent ineligible, holding that the "patent claims at issue here effectively claim the underlying laws of nature themselves."9 The Court reiterated that "laws of nature, natural phenomena, and abstract ideas" were not patent eligible.10 Prometheus made it more difficult to tell when a diagnostic method claim becomes patent eligible, and at the same time set a higher bar for patent eligibility.11 The Prometheus decision requires new strategies for drafting and prosecuting diagnostic method patent applications. The older strategies of relying on broadly drafted claims are unlikely to work. On those occasions when they do work, if the resulting patent has commercial value, the broad claims are likely to face post-grant challenge in the United States Patent and Trademark Office (USPTO) or the courts. Below we discuss possible strategies for maximizing claim scope to protect eligible subject matter after Prometheus. These strategies are equally applicable to drafting new applications, prosecuting already filed applications, and responding to post-grant challenges to issued patent claims.12 Strategy 1: Patent in Bite-Sized Chunks to Avoid Preemption Challenges A primary concern of the Prometheus Court was preemption—the idea that a patent claim could preempt future (or all) uses of a law of nature (e.g., a newly discovered correlation between a biomarker and the presence or absence of a disease). "[Court precedents] warn us against upholding patents that claim processes that too broadly preempt the use of a natural law."13 Before Prometheus, diagnostic method claims were often broadly written to cover what was legitimately believed to be an applicant's contribution and invention. The pre-Prometheus approach to claiming is shown graphically below, with the pie chart representing the intellectual property space covering all uses of the invention.  An approach preempting all uses of a law of nature with one broad claim is unlikely to succeed following Prometheus. But the same, or approximately the same, result can be achieved using a series of claims, with each claim having an intermediate scope that covers some applications of a law of nature. The post-Prometheus claiming approach is depicted below, with each pie "widget" representing a claim of intermediate scope. Collectively, all of the widgets can cover the same space as a simple, pre-Prometheus broad claim.  Thus, while a single broad claim that ties up all or almost all future uses of a law of nature is now unlikely to be granted (and, if granted, may not survive a post-grant challenge), it is possible to obtain several claims that in aggregate cover the same scope of subject matter as a single broad claim. This is a preferred post-Prometheus approach to patent application drafting and prosecution. If a claim is of intermediate scope and does not preempt all uses of a law of nature, a best practice is to point this out to the examiner in arguing against a patent-eligible subject matter rejection. For example, a claim may require detection of a biomarker using fewer than all available detecting methods. Pointing this out to the examiner is a strong argument against broad preemption and at the same time is an equally solid argument for patent eligibility. Furthermore, under the new America Invents Act, broad claims covering only the law of nature can be poisonous because they are unlikely to issue as patent claims. In order to meet patent eligibility, drafters need to disclose putative or definite applications of the law of nature. Strategy 2: Interview Each Application with a Patent Eligibility Rejection (i.e., a 35 U.S.C. § 101 Rejection) An elastic middle ground exists between the broad ambit of 35 U.S.C. § 10114 and subject matter that is patent ineligible (e.g., "laws of nature, natural phenomena, abstract ideas").15 In each case, the elastic line between patent ineligibility and patent eligibility is determined by a patent examiner, generally in conjunction with his or her supervisor, and often with consultation of a Technology Center Specialist. Interviews can favorably shift the elastic line, resulting in a broader claim. Thus, interviewing each case with a patent eligibility rejection is recommended when confronted with a Section 101 rejection.16 Strategy 3: Draw Parallels Between a Diagnostic Method Claim and Allowable Claims as Recited in the USPTO Guidelines17 In July 2012, the USPTO published interim guidelines18 to aid patent examiners in determining whether process claims (e.g., diagnostic method claims) that employ a law of nature are patent eligible. Pages 10 and 11 of the guidelines, for example, provide exemplary claims that the USPTO has determined are drawn to patent-eligible subject matter. When responding to a patent ineligibility rejection, consider pointing out the parallels between pending claims and one or more claims deemed patent eligible by the guidelines. These arguments by analogy may be the most powerful in convincing a patent examiner that a claim is patent eligible. Strategy 4: If Applicable, Argue That Your Claim Passes the Machine or Transformation Test Some claims may be performed using a particular machine or may transform matter to a different state or thing. For example, a diagnostic method claim may employ a novel antibody to detect a biomarker through the binding of the novel antibody to the biomarker, and may also employ a particular machine (e.g., a spectrophotometer) to detect the binding of the novel antibody to the biomarker. In Mayo Collaborative Servs. v. Prometheus Labs., Inc., 123 S. Ct. 1289, 1296 (2012), the Supreme Court clarified that the "'machine or transformation test' [while] not a definitive test of patent eligibility . . . is . . . an important and useful clue." Thus, if a claim uses a particular machine or transforms matter to a different state or thing, point out this "important and useful clue" to the examiner. As previously discussed, this argument can also be utilized in support of the contention that the claims do not preempt the entire "law of nature." Strategy 5: Argue Novelty (Both Generally and with Regard to Any Novel Claim Elements) and Non-Obviousness Normally, the satisfaction of different patent statute requirements is determined individually (e.g., satisfaction of the written description requirement under 35 U.S.C. § 112, 1st ¶ is determined separate from the non-obviousness requirement under 35 U.S.C. § 103). However, recent trends have blurred the lines between the statutory requirements such that one statutory requirement can determine in whole or in part whether another statutory requirement is met. For example, the description in a patent specification (e.g., 35 U.S.C. § 112, 1st ¶) can determine the scope of a means-plus-function or step-plus-function claim and whether or not the claim is indefinite (e.g., under Section 112, 2nd ¶), thereby influencing both Section 112, 2nd ¶ and Section 112, 6th ¶. Recently, the presence or absence of critical, superior, and unexpected results was held to influence whether or not claims including a narrow numerical range are anticipated by prior art disclosing a broader range that encompasses the narrow numerical range.19 Thus, for claims with numerical ranges, factors normally associated with obviousness can determine whether or not the claim is anticipated. Prometheus expands this trend because under the Court's decision, novelty (e.g., 35 U.S.C. § 102) and non-obviousness (e.g., 35 U.S.C. § 103) can influence whether or not a claim is drawn to patent-eligible subject matter. The Prometheus Court was concerned that claims drawn to a natural law "also contain other elements or a combination of elements, sometimes referred to as an 'inventive concept,' sufficient to ensure that the patent in practice amounts to significantly more than a patent upon the natural law itself."20 By "inventive concept," the Court appears to mean some combination of novelty and non-obviousness. For example, the Court determined that Prometheus' claims were not patent eligible, in part because "the steps in the claimed processes (apart from the natural laws themselves) involve well-understood, routine, conventional activity previously engaged in by researchers in the field"21 (i.e., they were not novel). The Court further determined that patentable subject matter may include a process that integrates a natural correlation by using one or more steps not "obvious, already in use, or purely conventional."22 In the coming years, the USPTO and the courts will likely define the relationship of Sections 102 and 103 to Section 101. Thus, the presence or absence of novelty (and non-obviousness) can influence whether a claim is patent eligible. Novelty (and non-obviousness) can therefore be advantageously used to argue for patent eligibility. For example, if there are specific novel claim elements (e.g., a novel biomarker or novel antibody to detect the biomarker), argue these as distinguishing the claim from the unpatentable claims in Prometheus. Also, argue non-obviousness, where appropriate, as a factor indicating patent subject matter eligibility. Strategy 6: Argue the Broad Ambit of Section 101 The Supreme Court previously affirmed the broad ambit of Section 101 patent-eligible subject matter: "In choosing such expansive terms . . . modified by the comprehensive 'any,' Congress plainly contemplated that the patent laws would be given wide scope." (Bilski v. Kappos, 130 S. Ct. 3218, 3225 (2010); citation omitted; emphasis added.) "Congress took this permissive approach to patent eligibility to ensure that 'ingenuity should receive a liberal encouragement.'" (Id.; citations omitted; emphasis added.) Based on the Supreme Court's broad interpretation of the scope of Section 101, issuance of a Section 101 rejection should be rare and the barrier to showing patent eligibility (e.g., to overcome a Section 101 rejection) should be low. This favors patent eligibility and, as a best practice, should generally always be argued. Strategy 7: Argue Integration of the Law of Nature The Prometheus Court, in its analysis, cited an earlier case that made use of a mathematical equation to determine when a rubber article was sufficiently cured. Although the mathematical equation by itself was not patent eligible, "the overall process was patent eligible because of the way the additional steps of the process integrated the equation into the process as a whole."23 Thus, it is important that a diagnostic method patent claim integrate the law of nature by including one or more steps, in addition to the natural law, in the diagnostic method claim. The one or more steps should contain at least one physical step (e.g., detecting a biomarker by binding the antibody to the biomarker) to demonstrate integration of the natural law. All integrative steps should generally be pointed out to a patent examiner when responding to a patent ineligibility reaction. Strategy 8: Consider Adding an Administering Step After a Determining Step The claims at issue in Prometheus did not require acting on the results of the diagnostic testing. Thus, if the level of 6-thioguanine was determined to be too low or too high, there was no requirement to adjust the 6-thioguanine dose in subsequent administration(s) of the drug. Requiring action after a positive diagnosis using the diagnostic method, such as administering a drug to treat the diagnosed disease or condition, may differentiate a claim from the unpatentable claims in Prometheus. If drugs other than the administered drug are available to treat the disease or condition, then the administering step also further ensures that the claim does not preempt the law of nature. If the drug being administered is novel, then the novelty is another element distinguishing the claim from Prometheus' patent-ineligible claims. Strategy 9: Stack Arguments to Tip the Balance to Patent Eligibility Because the line between patent ineligibility and patent eligibility is elastic, one way to show patent eligibility is by using a balance analogy. Analogous to adding weights to a balance pan to tip the balance, stacking arguments in favor of patent eligibility can tip a claim from patent ineligible to patent eligible. Arguments, as outlined above, can include the following:
As previously discussed, the best time to initially make these arguments is when interviewing the case. Conclusion Prometheus necessitates new strategies for drafting, prosecuting, and defending diagnostic method claims. After Prometheus, Section 101 rejections have increased in frequency. Many otherwise allowable diagnostic method claims are being rejected in new non-final Office Actions, with the Section 101 rejections the only rejections standing in the way of patentability. Additionally, it is estimated that more than three quarters of diagnostic method claims issued before Prometheus may now be patent ineligible and open to post-grant challenges. It would be prudent to analyze granted diagnostic method claims now and prepare for possible future post-grant challenges. Based on our experience prosecuting diagnostic methods claims after Prometheus, the strategies outlined above are useful for maximizing patent claim scope and patent protection. The strategies are equally applicable to drafting new applications, prosecuting already filed applications, and responding to post-grant challenges to issued patent claims.
1 A correlative diagnostic method claim may correlate a biomarker, a drug, a metabolite, an analyte level, a genetic polymorphism, or a genetic expression profile with a disease diagnosis, a disease prognosis, the presence or absence of a disease-inducing condition, a phenotype expression, actual or potential responsiveness of a drug, or actual or potential side effects from using a drug, in an individual patient.
wherein the level of 6-thioguanine less than about 230 pmol per 8x108 red blood cells indicates a need to increase the amount of said drug subsequently administered to said subject and
wherein the level of 6-thioguanine greater than about 400 pmol per 8x108 red blood cells indicates a need to decrease the amount of said drug subsequently administered to said subject.
6 See "Thioguanine," available at http://en.wikipedia.org/wiki/Tioguanine. 7 The therapeutic window of a drug is the range of drug dosages that can treat disease effectively while staying within the safety range; definition available at http://en.wikipedia.org/wiki/Therapeutic_window. 8 Curiously, the Court, for purposes of legal analyses, did not distinguish between specific, narrowly applicable correlations and broadly applicable natural principles (e.g., E = MC2) that are based on empirical observation. Many scientists, for example, would argue that laws of nature are by definition broadly applicable natural principles and that there are relatively few laws of nature (e.g., first law of thermodynamics, law of gravity, E=MC2). 9 Mayo Collaborative Servs. v. Prometheus Labs., Inc., 132 S. Ct. 1289, 1305 (2012). 10 Id. at 1293. Citations omitted. 11 We have recently seen a trend where otherwise allowable diagnostic method claims have, following Prometheus, been issued non-final Office Actions where, in each case, the only rejection is a 35 U.S.C. § 101 rejection. 12 One study estimates that many diagnostic method claims in granted patents may, following Prometheus, be patent ineligible. See E. J. Haanes and J. M. Canaves, "Stealing Fire: A Retrospective Survey of Biotech Patent Claims in the Wake of Mayo v. Prometheus," Nature Biotechnology 30(8): 758-760 (2012). 13 Mayo Collaborative Servs. v. Prometheus Labs., Inc., 132 S. Ct. 1289, 1294 (2012). 14 35 U.S.C. § 101 recites four broad categories as being patent-eligible subject matter: processes, machines, manufactures, and compositions of matter. New and useful improvements of these are patent eligible as well. 15 Mayo Collaborative Servs. v. Prometheus Labs., Inc., 132 S. Ct. 1289, 1294 (2012) (citations omitted). 16 When interviewing cases, we recommend having a decision maker present. Because of the newness of Prometheus, and the uncertainty about when a diagnostic method claim becomes patent eligible, some examiners are understandably reluctant to "make the call" regarding whether a proposed claim is patent eligible. Thus, having a supervisory patent examiner present during the interview can be helpful to furthering prosecution. 17 United States Patent and Trademark Office's "2012 Interim Procedure for Subject Matter Eligibility Analysis of Process Claims Involving Laws of Nature," July 3, 2012, available at: http://www.uspto.gov/patents/law/exam/2012_interim_guidance.pdf. 18 Id. 19 See Clearvalue, Inc. v. Pearl River Polymers, 560 F.3d 1291 (2009). 20 Mayo Collaborative Servs. v. Prometheus Labs., Inc., 132 S. Ct. 1289, 1294 (2012) (citations omitted). 21 Id. 22 Id. at 1299. 23 Id. at 1298. Recent Life Sciences Highlights MAQUET Cardiovascular Acquires LAAx Class Action Settlement Approved in Medicare Improvement Standard Case
Wilson Sonsini Goodrich & Rosati's Medical Device Conference
Phoenix 2013: The Medical Device and Diagnostic Conference for CEOs
Click here for a printable version of The Life Sciences Report This communication is provided for your information only and is not intended to © 2013 Wilson Sonsini Goodrich & Rosati, Professional Corporation |






 Ed Poplawski is a leader of Wilson Sonsini Goodrich & Rosati's intellectual property litigation practice, with a primary focus on patent litigation. Nationally regarded as one of the nation's preeminent trial lawyers, he headed Sidley Austin's West Coast IP practice and served as global co-chair of that firm's IP litigation group prior to joining WSGR. Ed earned his J.D. from the Villanova University School of Law in 1983, and holds a master's degree and a bachelor of science degree in mechanical engineering from Drexel University.
Ed Poplawski is a leader of Wilson Sonsini Goodrich & Rosati's intellectual property litigation practice, with a primary focus on patent litigation. Nationally regarded as one of the nation's preeminent trial lawyers, he headed Sidley Austin's West Coast IP practice and served as global co-chair of that firm's IP litigation group prior to joining WSGR. Ed earned his J.D. from the Villanova University School of Law in 1983, and holds a master's degree and a bachelor of science degree in mechanical engineering from Drexel University. Vera Elson has substantial bench and jury trial experience with complex, high-stakes cases in federal district courts across the country and before the ITC relating to matters such as patents, trade secrets, and copyright claims. She has successfully handled cases for marquee clients such as GE Healthcare and Seagate Technology. Vera also regularly provides strategic IP counseling for technology clients. Before practicing law, she worked as an integrated-circuit designer. Vera received her J.D. from USC's Gould School of Law in 1991 and holds an M.S. in engineering from UCLA.
Vera Elson has substantial bench and jury trial experience with complex, high-stakes cases in federal district courts across the country and before the ITC relating to matters such as patents, trade secrets, and copyright claims. She has successfully handled cases for marquee clients such as GE Healthcare and Seagate Technology. Vera also regularly provides strategic IP counseling for technology clients. Before practicing law, she worked as an integrated-circuit designer. Vera received her J.D. from USC's Gould School of Law in 1991 and holds an M.S. in engineering from UCLA.  Paul Tripodi II focuses on complex patent litigation and trade secret misappropriation spanning a broad range of technologies, including pharmaceuticals, medical devices, smartphones, wireless navigation, integrated circuits, data transmission, and video games. He has been involved in both bench and jury trials, as well as proceedings before international tribunals and the International Trade Commission. Paul received his J.D. from the UCLA School of Law in 1992, an M.S. in chemical physics from the California Institute of Technology in 1989, and a B.S. in chemistry from Pennsylvania State University in 1987.
Paul Tripodi II focuses on complex patent litigation and trade secret misappropriation spanning a broad range of technologies, including pharmaceuticals, medical devices, smartphones, wireless navigation, integrated circuits, data transmission, and video games. He has been involved in both bench and jury trials, as well as proceedings before international tribunals and the International Trade Commission. Paul received his J.D. from the UCLA School of Law in 1992, an M.S. in chemical physics from the California Institute of Technology in 1989, and a B.S. in chemistry from Pennsylvania State University in 1987. Olivia Kim specializes in complex patent infringement litigation in areas such as biometric sensors, biotechnology, computer processor architecture, graphical user interfaces, Internet payment systems, mobile phones, network security, pharmaceuticals, and plasma display panels. Experienced in all stages of litigation, Olivia has participated in a significant number of jury and bench trials. Before becoming an attorney, she was a researcher at UCLA's immunology and physiology labs. Fluent in Korean, Olivia received a J.D. from the University of Southern California Gould School of Law in 2003 and a B.S. in biochemistry from UCLA in 1998.
Olivia Kim specializes in complex patent infringement litigation in areas such as biometric sensors, biotechnology, computer processor architecture, graphical user interfaces, Internet payment systems, mobile phones, network security, pharmaceuticals, and plasma display panels. Experienced in all stages of litigation, Olivia has participated in a significant number of jury and bench trials. Before becoming an attorney, she was a researcher at UCLA's immunology and physiology labs. Fluent in Korean, Olivia received a J.D. from the University of Southern California Gould School of Law in 2003 and a B.S. in biochemistry from UCLA in 1998. Sandra Fujiyama represents clients in industries ranging from biotechnology to electronics, with a particular emphasis on patent litigation. An experienced trial lawyer, she also has expertise in general IP counseling, prosecuting patent and trademark applications, performing trademark registerability analyses, and conducting IP due diligence. Sandra earned her J.D. from the UCLA School of Law in 1998 and a B.S. in biochemistry from UCLA in 1995.
Sandra Fujiyama represents clients in industries ranging from biotechnology to electronics, with a particular emphasis on patent litigation. An experienced trial lawyer, she also has expertise in general IP counseling, prosecuting patent and trademark applications, performing trademark registerability analyses, and conducting IP due diligence. Sandra earned her J.D. from the UCLA School of Law in 1998 and a B.S. in biochemistry from UCLA in 1995.




